
Lovastatine en ALD
New England Journal of Medicine 2010 Jan 21;362(3):276-277.
 In 1998 werd in het New England Journal of Medicine gerapporteerd dat het cholesterol verlagende medicijn lovastatine zeer langketen vetzuren (ZLKV) kon verlagen in het bloed van patiënten met X-gebonden adrenoleukodystrofie (ALD). Sindsdien zijn er veel mensen met ALD, vooral in de V.S., die dit middel gebruiken in de hoop dat het het ziektebeloop gunstig zal beïnvloeden. De daaropvolgende jaren echter ontstonden er toch twijfels over de effectiviteit van lovastatine: andere onderzoekers vonden geen effect op ZLKV bij behandeling van muizen met ALD en een onderzoek met simvastatine (een stof die erg lijkt op lovastatine) bij kinderen met X-ALD liet ook geen effect zien op de ZLKV in het bloed.
In 1998 werd in het New England Journal of Medicine gerapporteerd dat het cholesterol verlagende medicijn lovastatine zeer langketen vetzuren (ZLKV) kon verlagen in het bloed van patiënten met X-gebonden adrenoleukodystrofie (ALD). Sindsdien zijn er veel mensen met ALD, vooral in de V.S., die dit middel gebruiken in de hoop dat het het ziektebeloop gunstig zal beïnvloeden. De daaropvolgende jaren echter ontstonden er toch twijfels over de effectiviteit van lovastatine: andere onderzoekers vonden geen effect op ZLKV bij behandeling van muizen met ALD en een onderzoek met simvastatine (een stof die erg lijkt op lovastatine) bij kinderen met X-ALD liet ook geen effect zien op de ZLKV in het bloed.
De afgelopen jaren is in het AMC in Amsterdam een onderzoek uitgevoerd om deze twijfels weg te nemen en vast te stellen of lovastatine daadwerkelijk de ZLKV in het bloed van ALD patiënten kan verlagen. Aan dit onderzoek deden 14 mannen met ALD mee. Zij zijn gedurende 6 maanden behandeld met lovastatine en daarna 6 maanden met een placebo (“nepmedicijn”), of eerste placebo en dan lovastatine. Zowel de deelnemers als de onderzoekers wisten niet wat er op welk moment gebruikt werd tijdens de onderzoeksperiode (“dubbelblind”). Na de behandelingsperiode werden alle bloedmonsters en data geanalyseerd.
Zoals verwacht resulteerde de behandeling met lovastatine in een verlaging van het cholesterol. Daarnaast bleek lovastatine een 20% verlaging van ZLKV in het bloed te veroorzaken. Echter, deze waarden waren nog steeds 2 tot 3 maal hoger dan normaal. Bovendien was er in de witte en rode bloedcellen geen verlaging van ZLKV meetbaar na behandeling met lovastatine. ZLKV zijn niet wateroplosbaar en worden in het bloed getransporteerd in “vetdeeltjes” (zogenaamde lipoproteïnen). Ook in deze vetdeeltjes was er geen daling in ZLKV meetbaar na behandeling met lovastatine.
Dit onderzoek toont aan dat lovastatine geen echte daling van ZLKV bewerkstelligt, maar een pseudodaling van ZLKV. Lovastatine moet dan ook niet voorgeschreven worden als behandeling voor ALD.
Gentherapie voor ALD
Science. 2009 Nov 6;326(5954):818-23.
Het team van Dr Nathalie Cartier en Patrick Aubourg (Saint Vincent de Paul Ziekenhuis, Parijs, Frankrijk) heeft twee jongetjes van 7 jaar met vroege verschijnselen van de cerebrale vorm van adrenoleukodystrofie behandeld met gentherapie. De twee jongens kwamen in aanmerking voor een beenmergtransplantatie, maar er waren geen geschikte donoren.
Er werd beenmerg van de patiënten afgenomen. Hieruit werden in het laboratorium stamcellen geïsoleerd. Vervolgens werd het normale ALD gen in de cellen ingebracht m.b.v. een onschadelijk gemaakt HIV-achtig virus. De jongens ondergingen chemotherapie om te voorkomen dat hun beenmerg nog stamcellen kon aanmaken. Vervolgens werden de gecorrigeerde stamcellen terug gebracht.
De twee jongens werden gedurende 24 en 30 maanden vervolgd. Vijftien procent van de bloedcellen produceerde het normale ALD eiwit gedurende deze periode.
Herhaalde MRI scans van de hersenen en cognitieve tests toonden aan dat de progressie van de ziekte na 14 tot 16 maanden stopte. Dit is vergelijkbaar met het klinische beloop na een normale beenmergtransplantatie.
Foto Patrick Aubourg
Behandeling van ALD
Vanaf het begin van de 80-er jaren is op allerlei manieren geprobeerd iets aan ALD te doen. De meeste bekende behandeling is waarschijnlijk die met “Lorenzo’s olie”. Over deze behandeling bij een jongen met ALD, Lorenzo Odone is in de Verenigde Staten zelfs een speelfilm gemaakt (Lorenzo’s oil), die in 1993 in Nederland in première ging. Maar er zijn ook andere behandelingen geprobeerd. Hierover later meer.
Bij de behandeling van ALD dienen een aantal zaken goed van elkaar te worden onderscheiden.
Hormonale stoornissen
Bijnierschorsinsufficiëntie: Bekend is dat tenminste 75% van alle jongens en mannen met ALD een bijnierschorsinsufficiëntie heeft. Onbehandeld kan dit leiden tot ernstige ziekte, en – soms zelfs – plotseling overlijden. Behandeling is makkelijk, en bestaat uit het in tabletvorm geven van de hormonen waaraan een tekort is. Meestal wordt dan hydrocortison gegeven, soms wordt daarnaast nog fludrocortison gegeven. De laatste jaren is ontdekt dat ook een ander hormoon, dehydroepiandrosteron (DHEA), vaak niet in voldoende mate gemaakt wordt door de bijnieren. Het is onduidelijk of dit oorzaak of gevolg is. Bij andere ziekten waarbij de bijnierschorsfunctie gestoord is, en ook het DHEA in bloed verlaagd is, wordt door sommige artsen ook dit hormoon in tabletvorm voorgeschreven.
Onvoldoende mannelijk geslachtshormoon: Onvoldoende functie van de mannelijke geslachtsklieren, de testikels, wordt ook bij zo’n 75% van de patiënten gezien. Dit kan soms alleen door laboratoriumonderzoek aangetoond worden. Indien er te weinig mannelijk geslachtshormoon wordt geproduceerd, en er klachten zijn van afgenomen spiermassa, minder zin in sex, en impotentie, dan kan overwogen worden ook dit hormoon in tabletvorm (of met injectie) aan te vullen. Zijn er problemen met de erectie zonder dat daarvoor een hormoontekort verantwoordelijk lijkt, dan kan het gebruik van sildenafil (Viagra®) worden overwogen. Gebruik van dit middel kan echter voor sommige mensen (bijvoorbeeld hartpatiënten) zeer schadelijk zijn.
Schildklierziekten: Een stoornis van de functie van de schildklier bij ALD is beschreven, maar omdat schildklierziekten erg veel voorkomen is het niet zeker of dit door X-ALD wordt veroorzaakt. Onderzoek naar een te traag werkende schildklier dient te gebeuren bij klachten van extreme vermoeidheid, gewichtstoename, obstipatie, depressie en haaruitval.
Neurologische problemen
Hierbij dienen de snel progressieve hersenbeschadiging zoals die bij cerebrale ALD voorkomt en de ruggenmerg- en zenuwproblemen van AMN van elkaar te worden onderscheiden. Het bewegingsapparaat (gewrichten, spieren) kan als gevolg van de afwijkingen aan het zenuwstelsel worden overbelast of beschadigd.
Cerebrale ALD
Momenteel is er maar één behandeling die bij cerebrale ALD kan helpen: beenmergtransplantatie. Voorwaarde is dan wel dat de uitvalsverschijnselen licht zijn. Daarnaast moet er een geschikte donor zijn, bij voorkeur een gezonde broer of zus. Het is een risicovolle ingreep die gepaard gaat met een zware behandeling (chemotherapie) en een isolatie van tenminste een maand (vanwege het infectiegevaar) inhoudt. Ook is behandeling met afstotingsremmende middelen nodig. Het werkingsmechanisme is niet geheel bekend. Aangenomen wordt dat de cellen van het beenmerg van de donor in staat zijn in het lichaam van de ontvanger ZLKV af te breken, zodat er minder gestapeld wordt. Een andere theorie is dat door de behandeling met cytostatica het afweersysteem dat de ontstekingsreactie in de hersenen veroorzaakt wordt stilgelegd. Het risico op complicaties van deze behandeling neemt toe naarmate een patiënt ernstiger is aangedaan, hij ouder is, en een donor heeft die minder goed bij hem past. Voor alle duidelijkheid: een transplantatie moet alleen overwogen worden bij het begin van hersenbeschadiging, en niet bij AMN waarbij alleen het ruggenmerg en zenuwen in armen en benen zijn betrokken.
Dan zijn er een aantal symptomen bij ALD die behandeld kunnen worden. Bij cerebrale ALD en de andere vormen met ernstige hersenbeschadiging kunnen epileptische aanvallen ontstaan, die als zij vaak voorkomen of zeer hinderlijk zijn moeten worden behandeld. Dit kan met de voor andere epilepsieën gebruikte middelen, zoals o.a. valproaat (Depakine®) of carbamazepine (Tegretol®). Vanwege voortschrijdende afwijkingen aan de hersenen kunnen slikproblemen ontstaan. Eten en drinken worden daardoor steeds moeilijker, en verslikken (met alle gevolgen van dien) is een gevaarlijk probleem. Dit kan worden opgelost via een maag- of duodenumsonde, en eventueel met een zogenaamde PEG (percutane endoscopische gastrostomie) sonde waarmee een slangetje door de buikwand heen direct in de maag wordt gelegd.
AMN
Bij mannen met AMN en draagsters met AMN-achtige symptomen staan andere verschijnselen op de voorgrond. Spasticiteit van de benen kan een zeer hinderlijk en pijnlijk probleem worden. Met bepaalde spierverslappende middelen (waaronder baclofen (Lioresal®), dantroleen (Dantrium®), en tizanidine (Sirdalud®) kan hier iets tegen worden gedaan. Soms zijn de klachten zo hevig dat met een pompje continu baclofen direct in de ruggenmergsholte kan worden gegeven. Incontinentie voor urine is een ander groot probleem. Er ontstaat een probleem tussen de samenwerking van de sluitspier van de blaas, en de spieren in de blaaswand. Soms is de blaasspier te sterk aangespannen en kan de urine de blaas niet verlaten, soms zijn de spieren in de blaaswand zo sterk aangespannen dat de blaas zich voortdurend ledigt. Onderzoek door een uroloog is dan nodig om uit te maken wat het probleem is, en te kijken of er met bepaalde medicijnen iets tegen te doen is. Een probleem waar veel minder over gesproken wordt is de incontinentie voor ontlasting. Het is een groot probleem dat kan leiden tot een sociaal isolement. Incontinentie voor ontlasting kan ook andere oorzaken hebben, en daarom is het belangrijk dat dat wordt uitgezocht door een maag-darm-lever arts (gastro-enteroloog). Mocht er ondanks het inroepen van de hulp van deze specialisten geen verbetering zijn, dan is het goed een incontinentieverpleegkundige te raadplegen. De meeste grote ziekenhuizen hebben zo’n verpleegkundige.
Het bewegingsapparaat: Tenslotte kan er als gevolg van de spasticiteit een overbelasting van de wervelkolom (rug en nek), en van de heupen, knieën en enkels ontstaan. Met tegen gewrichtsklachten gerichte pijnbestrijders zoals onder andere ibuprofen (Brufen®) en diclofenac (Voltaren®) is hier meestal wel iets tegen te doen.
Experimentele therapieën
Algemeen: Van veel middelen is inmiddels duidelijk dat zij niet of nauwelijks werken. Behandeling met prednison, gamma-globuline (immunoglobuline), bepaalde cytostatica en ß-interferon hielp niet. Enig effect van een vetbeperkt dieet dat wordt aangevuld met “Lorenzo’s olie” (een combinatie van erucazuur en oliezuur) is niet uitgesloten bij patiënten die nog geen neurologische uitvalsverschijnselen hebben, maar bij patiënten met neurologische klachten is geen aantoonbaar effect gemeld.
“Lorenzo’s olie”
Lorenzo’s olie therapie is gebaseerd op de kennis dat ZLKV niet alleen via ons voedsel binnenkomen maar ook door het lichaam zelf geproduceerd worden d.m.v. ketenverlenging van lang-keten vetzuren. Laboratorium experimenten lieten zien dat het kweken van fibroblasten van ALD patiënten in de aanwezigheid van onverzadigde vetzuren resulteerde in verlaging van de ZLKV. Behandeling van ALD patiënten met een vetbeperkt dieet dat wordt aangevuld met “Lorenzo’s olie” (een combinatie van erucazuur en oliezuur) resulteerde in een normale concentratie ZLKV in het bloed. Deze spectaculaire en hoopgevende resultaten hebben ertoe geleid dat er wereldwijd honderden ALD patiënten behandeld werden met “Lorenzo’s olie”. Echter, al snel bleek dat de olie geen effect had op het ziektebeloop. “Lorenzo’s olie” kan ALD patiënten niet beschermen tegen het krijgen van AMN of cerebrale ALD. Patiënten die al neurologische symptomen hadden verslechterden, ondanks de normalisatie van de ZLKV in het plasma. Er zijn vele gevallen bekend van patiënten die bij aanvang van de therapie nog geen symptomen hadden, maar ondanks de olie toch symptomen hebben gekregen. Onderzoek in de hersenen van overleden patiënten die de olie gebruikt hadden wees uit dat de olie geen effect heeft op de ZLKV in de hersenen, de ZLKV waarden waren niet verlaagd. Men concludeerde dat “Lorenzo’s olie” de hersenen niet bereikt, hetgeen wordt veroorzaakt door de bloed-hersen barrière. De bloed-hersen barrière scheidt de hersenen van de rest van het lichaam en het beschermt de hersenen tegen het binnendringen van allerlei mogelijk schadelijke stoffen die in het bloed kunnen voorkomen.
Neurologisch asymptomatische jongens jonger dan 10 jaar die met “Lorenzo’s olie” worden behandeld krijgen volgens een in Amerika uitgevoerd retrospectief onderzoek bij grote therapietrouw minder vaak cerebrale afwijkingen dan de jongens die de olie minder goed gebruikten. Daarom wordt het gebruik van “Lorenzo’s olie” in Amerika wel aangeprezen voor neurologisch asymptomatische jongens.
In 2010, 21 jaar na het ontwikkelen van “Lorenzo’s olie” is het eigenlijk nog steeds niet goed bekend wat nu werkelijk het effect van “Lorenzo’s olie” is op het ziekte beloop. De retrospectieve studie hierboven beschreven is misschien hoopgevend, maar enkele kritische kanttekeningen moeten wel geplaatst worden. De belangrijkste is het ontbreken van de controle (onbehandelde) groep.
Op basis van de bevindingen is men in Baltimore (VS) in 2007 begonnen met een groot onderzoek naar het effect van “Lorenzo’s olie” bij ALD patiënten. De opzet is een echte studie met een onbehandelde (placebo) groep.
Lovastatine
In 1998 werd door de groep van Dr. Inderjit Singh aangetoond dat behandeling van fibroblasten van ALD patiënten met lovastatine resulteert in een verlaging van de ZLKV in deze cellen. Lovastatine is een cholesterol verlagend middel dat door vele mensen buiten Nederland, met te hoge cholesterol waarden in het bloed, gebruikt wordt. In Nederland is het middel niet geregistreerd en niet verkrijgbaar. Het middel heeft vrijwel geen bijwerkingen. De onderzoekers behandelde ook een kleine groep ALD patiënten. Binnen enkele maanden waren de ZLKV in het bloed genormaliseerd in een aantal patiënten. Over het effect op de concentraties ZLKV in de hersenen is niets bekend, daar zal nog meer onderzoek voor nodig zijn. Het mechanisme waarmee lovastatine de ZLKV verlaagt, is ook nog niet opgehelderd. Daarnaast zijn er enkele zaken bij deze studie onduidelijk.
In 2007 is er in het Academisch Medisch Centrum een studie uitgevoerd om het effect van lovastatine goed in kaart te brengen. Veertien ALD patiënten namen deel aan de studie. De studie is opgezet als een dubbelblinde placebo-gecontroleerde cross-over studie. Dit betekent dat de patiënten gedurende de helft van de behandelingsperiode een placebo krijgen en gedurende de andere helft lovastatine. Alleen weet zowel de patiënt als de behandelend arts niet wat de patiënt in welke periode krijgt. De resultaten van de studie zijn in januari 2010 gepubliceerd in de New England Journal of Medicine. De behandeling met lovastatine resulteerde in een verlaging van het plasma cholesterol. De ZLKV daalde met ongeveer 20%, maar bleven nog steeds 2-3x hoger dan het controle niveau. Er was echter geen effect van de behandeling met lovastatine op de ZLKV niveaus in bloedcellen.
Gentherapie
Dr Patrick Aubourg en Dr Nathalie Cartier zijn in 1994 begonnen met het ontwikkelen van gentherapie voor ALD. De procedure is een ex vivo benadering. Dit houdt in dat de beenmergcellen van de patiënt geïsoleerd worden, vervolgens worden de beenmergcellen in het laboratorium behandeld met een onschadelijk virus waarin een normale kopie van het ALD gen aangebracht is. Na behandeling worden de beenmergcellen van de patiënt, die nu een normaal ALD eiwit produceren, weer teruggegeven aan de patiënt. In 2006 is de eerste ALD patiënt behandeld, in 2007 de tweede en in 2008 nog een derde patiënt. Alle drie waren jongens met vroege tekenen van witte stof veranderingen. De resultaten van de behandeling van de eerste twee getransplanteerde jongens zijn in 2009 gepubliceerd in Science.
De twee jongens werden gedurende 24 en 30 maanden vervolgd. Vijftien procent van de bloedcellen produceerde het normale ALD eiwit gedurende deze periode. Herhaalde MRI scans van de hersenen en cognitieve tests toonden aan dat de progressie van de ziekte na 14 tot 16 maanden stopte. Dit is vergelijkbaar met het klinische beloop na een normale beenmergtransplantatie.
Tot slot
Wanneer bij iemand de diagnose ALD is gesteld kan dit tot ingrijpende veranderingen in een gezin of familie leiden. Maar hoe moeilijk het ook is hiermee om te gaan, er mag nooit en te nimmer worden nagelaten onderzoek te doen bij de overige familieleden, als zij dat tenminste goed vinden. In de meeste gevallen blijken er namelijk meer gevallen per familie te zijn. En vroege herkenning van patiënten is noodzakelijk om vroegtijdig een bijnierschorsinsufficiëntie te ontdekken en te behandelen, of te gaan zoeken naar een geschikte donor voor een beenmergtransplantatie.
Wanneer de diagnose is gesteld is het belangrijk om in contact te blijven met een specialist die iets af weet van ALD. Zo kan regelmatig worden onderzocht of er een bijnierschorsinsufficiëntie is ontstaan, en of er afwijkingen in de hersenen zijn die een beenmergtransplantatie noodzakelijk maken.
Heel belangrijk is het om in contact te blijven met lotgenoten en medepatiënten, die sinds 1993 verenigd zijn in “De Belangenvereniging X-gebonden Adrenoleukodystrofie”. Deze vereniging heeft medische adviseurs die regelmatig in contact zijn met de beste onderzoekers op ALD gebied in binnen- en buitenland. Het motto van de Amerikaanse vereniging van patiënten met witte stof ziekten, de United Leukodystrophy Foundation (ULF), is overigens niet voor niets: “You are not alone …..”.
Feiten over ALD
X-chromosoom gebonden adrenoleukodystrofie (ALD) is een erfelijke metabole ziekte waarbij schade optreedt aan bijnieren, testis, hersenen en ruggenmerg. De ziekte werd voor het eerst beschreven in 1923 door de artsen Siemerling en Creutzfeldt als een gecombineerde bijnier- en hersenziekte. In 1970 werd de naam adrenoleukodystrofie geïntroduceerd; waarbij “adreno” verwijst naar de bijnieren, “leuko” verwijst naar de witte stof (het myeline, dit is een soort isolatielaag rondom de zenuwen) en “dystrofie” betekent beschadigd. In 1977 werd ontdekt dat er naast de hersenvorm van adrenoleukodystrofie ook een vorm is die zich openbaart in de volwassen leeftijd en waarbij het ruggenmerg aangetast raakt. Deze vorm wordt adrenomyeloneuropathy (AMN) genoemd: myelo verwijst naar het ruggenmerg en neuropathie verwijst naar het niet goed functioneren van één of meerdere zenuwen.
Auteurs: Dr. Wouter van Ballegoij (neuroloog in opleiding), Dr. Marc Engelen ((kinder)neuroloog) en Dr. Stephan Kemp (onderzoeker)
De biochemie van ALD
Adrenoleukodystrofie ontstaat bij ongeveer 1 op de 15.000 mensen. De ziekte wordt veroorzaakt door een afwijking in het ABCD1 gen. Dit gen produceert het adrenoleukodystrofie proteïne (ALDP). Het ALDP behoort tot een familie van transporteiwitten: eiwitten die bepaalde moleculen de cel (of een onderdeel van de cel) in- of uit transporteren. Het ALDP is verantwoordelijk voor het transport van verzadigde zeer langketen vetzuren (ZLKV) het peroxisoom in. Een van de functies van peroxisomen is het afbreken van ZLKV. Door het defect in het ALDP raakt het afbraakproces van de ZLKV ernstig gestoord, waardoor ZLKV stapelen in de cellen, weefsels en organen. Een te hoge concentratie ZLKV is schadelijk voor cellen. Maar waarom adrenoleukodystrofie patiënten specifiek klachten ontwikkelen aan testis, bijnieren, ruggenmerg en hersenen is nog niet opgehelderd. In het ruggenmerg leidt stapeling van ZLKV tot schade aan de zenuwen, waardoor onder andere de het lopen gestoord raakt. In de hersenen kan de stapeling leiden tot ernstige beschadiging van de witte stof (myeline); dit proces wordt demyelinisatie genoemd. In bijnieren resulteert de schade in het ontstaan van bijnierschorsinsufficiëntie.
 Figuur 1: ZLKV zijn vetzuren die bestaan uit meer dan 22 koolstofatomen. Vetzuren maken deel uit van plantaardige en dierlijke vetten en komen voor in ons dagelijks dieet. De vetzuren die stapelen in adrenoleukodystrofie worden echter voornamelijk door het lichaam zelf aangemaakt uit langketen vetzuren. Om de balans tussen aanmaak en afbraak te bewaken wordt het teveel aan ZLKV afgebroken in de peroxisomen. Iedere cel, met uitzondering van de rode bloedcellen, bevat peroxisomen. Door de mutatie in het ABCD1 gen en het hierdoor niet goed functionerende ALDP ontstaat er een defect in de afbraakroute. De onbalans tussen de ZLKV-aanmaak en -afbraak resulteert in de stapeling van ZLKV in cellen en weefsels.
Figuur 1: ZLKV zijn vetzuren die bestaan uit meer dan 22 koolstofatomen. Vetzuren maken deel uit van plantaardige en dierlijke vetten en komen voor in ons dagelijks dieet. De vetzuren die stapelen in adrenoleukodystrofie worden echter voornamelijk door het lichaam zelf aangemaakt uit langketen vetzuren. Om de balans tussen aanmaak en afbraak te bewaken wordt het teveel aan ZLKV afgebroken in de peroxisomen. Iedere cel, met uitzondering van de rode bloedcellen, bevat peroxisomen. Door de mutatie in het ABCD1 gen en het hierdoor niet goed functionerende ALDP ontstaat er een defect in de afbraakroute. De onbalans tussen de ZLKV-aanmaak en -afbraak resulteert in de stapeling van ZLKV in cellen en weefsels.
Epidemiologie
Adrenoleukodystrofie komt over de hele wereld voor en binnen alle culturen. Er zijn geen aanwijzingen dat er geografische verschillen zijn.
Genetica
Adrenoleukodystrofie is een X-chromosoom gebonden ziekte. Dit betekent dat het verantwoordelijke gen op het X-chromosoom ligt. Mannen hebben 1 X-chromosoom en 1 Y-chromosoom (XY; Figuur 2). Wanneer de vader drager is van het aangedane adrenoleukodystrofie gen, heeft hij geen gezond “back-up” adrenoleukodystrofie gen. Hij zal dus adrenoleukodystrofie-gerelateerde klachten ontwikkelen. Vrouwen hebben 2 X-chromosomen (XX; Figuur 2). Vrouwen die het defecte adrenoleukodystrofie gen dragen werden voorheen “draagsters” genoemd, omdat zij wel een gezond adrenoleukodystrofie gen hebben op het andere X-chromosoom. Vroeger dacht men dat de meeste draagsters weinig tot geen klachten ontwikkelen. Onderzoek in het AMC onder een grote groep “draagsters” heeft echter laten zien dat dit beeld niet klopt. Het is nu duidelijk dat tenminste 80% van de vrouwen met ALD klachten ontwikkelt (zie ook verderop). De term “draagsters” is dan ook misleidend en zou niet meer gebruikt moeten worden. Tegenwoordig spreken we over “vrouwen met ALD”. Waarom vrouwen in de regel iets lichtere klachten ontwikkelen dan mannen kan hoogstwaarschijnlijk verklaard worden door de aanwezigheid van het back-up gen op het tweede X-chromosoom. Mogelijk beschermt deze normale kopie vrouwen met ALD tegen het ontstaan van schade aan de hersenen en bijnieren.
 Figuur 2: (Links) Van een vrouw met ALD zal de helft van haar kinderen ook ALD krijgen. Een dochter krijgt 1 X-chromosoom van vader en 1 X-chromosoom van moeder. Zij heeft 50% kans het aangedane ALD gen te krijgen en 50% kans het gezonde ALD gen van haar moeder te krijgen. Een zoon krijgt zijn Y-chromosoom van vader en het X-chromosoom van moeder. Dus ook een zoon heeft 50% kans het aangedane ALD gen van zijn moeder te ontvangen. (Rechts) Voor een X-chromosoom gebonden ziekte als ALD geldt dat wanneer een aangedane vader kinderen krijgt zijn zonen nooit de ziekte krijgen. Hij geeft altijd zijn Y-chromosoom door aan zijn zonen en het X-chromosoom komt van moeder. Daarentegen zullen al zijn dochters het aangedane ALD gen ontvangen, omdat zij altijd zijn enige aangedane X-chromosoom ontvangen.
Figuur 2: (Links) Van een vrouw met ALD zal de helft van haar kinderen ook ALD krijgen. Een dochter krijgt 1 X-chromosoom van vader en 1 X-chromosoom van moeder. Zij heeft 50% kans het aangedane ALD gen te krijgen en 50% kans het gezonde ALD gen van haar moeder te krijgen. Een zoon krijgt zijn Y-chromosoom van vader en het X-chromosoom van moeder. Dus ook een zoon heeft 50% kans het aangedane ALD gen van zijn moeder te ontvangen. (Rechts) Voor een X-chromosoom gebonden ziekte als ALD geldt dat wanneer een aangedane vader kinderen krijgt zijn zonen nooit de ziekte krijgen. Hij geeft altijd zijn Y-chromosoom door aan zijn zonen en het X-chromosoom komt van moeder. Daarentegen zullen al zijn dochters het aangedane ALD gen ontvangen, omdat zij altijd zijn enige aangedane X-chromosoom ontvangen.
Het klinisch beloop
Patiënten met adrenoleukodystrofie zijn vrij van klachten bij geboorte.
Bij mannen is de eerste uiting van adrenoleukodystrofie meestal bijnierschorsinsufficiëntie. Dit kan al ontstaan in het eerste levensjaar, maar ook pas veel later. Bijna alle mannen en een groot deel van de vrouwen met ALD ontwikkelt als volwassene schade aan het ruggenmerg (ook wel myelopathie). Mannen met adrenoleukodystrofie kunnen een ontstekingsreactie in de hersenen ontwikkelen waarbij deze ernstig beschadigd raken. Deze vorm wordt cerebrale adrenoleukodystrofie (cerebrale ALD) genoemd. Cerebrale ALD kan de eerste uiting van adrenoleukodystrofie zijn of ontstaan in combinatie met bijnierschorsinsufficiëntie en / of myelopathie (Figuur 3). Vrouwen met ALD kunnen ook klachten ontwikkelen. Tenminste 80% van de aangedane vrouwen heeft op de leeftijd van 60 jaar klachten van ruggenmergschade (myelopathie). Vrouwen met ALD ontwikkelen vrijwel nooit bijnierschorsinsufficiëntie of cerebrale ALD.
 Figuur 3: Het klinische spectrum van ALD bij mannen. Patiënten met ALD vertonen geen symptomen bij geboorte. De gekleurde balken geven het leeftijdsbereik aan waarop klachten kunnen ontstaan voor bijnierschorsinsufficiëntie (donkerpaarse balk), myelopathie (licht paars) en cerebrale ALD (groene balk). De bijnierschorsinsufficiëntie kan al in het eerste levensjaar ontstaan. Als volwassene ontwikkelen mannen steevast ruggenmergschade (myelopathie). Cerebrale ALD kan op elke leeftijd ontstaan. De jongste patiënt ooit beschreven was 3 jaar oud. De stapeling van ZLKV in weefsels resulteert in bijnierschorsinsufficiëntie en schade aan het ruggenmerg (samen aangeduid als adrenomyeloneuropathie of AMN). Het ontstaan van cerebrale ALD wordt hoogstwaarschijnlijk bepaald door het samenspel van het primaire ALD gendefect en een combinatie van tot nu toe onbekende omgevingsfactoren en/of genetische factoren. Het is belangrijk om te beseffen dat patiënten met een bijnierschorsinsufficiëntie en/of myelopathie het risico lopen om alsnog cerebrale ALD te ontwikkelen.
Figuur 3: Het klinische spectrum van ALD bij mannen. Patiënten met ALD vertonen geen symptomen bij geboorte. De gekleurde balken geven het leeftijdsbereik aan waarop klachten kunnen ontstaan voor bijnierschorsinsufficiëntie (donkerpaarse balk), myelopathie (licht paars) en cerebrale ALD (groene balk). De bijnierschorsinsufficiëntie kan al in het eerste levensjaar ontstaan. Als volwassene ontwikkelen mannen steevast ruggenmergschade (myelopathie). Cerebrale ALD kan op elke leeftijd ontstaan. De jongste patiënt ooit beschreven was 3 jaar oud. De stapeling van ZLKV in weefsels resulteert in bijnierschorsinsufficiëntie en schade aan het ruggenmerg (samen aangeduid als adrenomyeloneuropathie of AMN). Het ontstaan van cerebrale ALD wordt hoogstwaarschijnlijk bepaald door het samenspel van het primaire ALD gendefect en een combinatie van tot nu toe onbekende omgevingsfactoren en/of genetische factoren. Het is belangrijk om te beseffen dat patiënten met een bijnierschorsinsufficiëntie en/of myelopathie het risico lopen om alsnog cerebrale ALD te ontwikkelen.
Bijnierschorsinsufficiëntie, of zelfs een levensbedreigende Addison-crisis, kan het eerste symptoom zijn van adrenoleukodystrofie bij jongens en mannen. Dit kan jaren of zelfs decennia voor het ontstaan van neurologische symptomen optreden. Een onderzoek met neurologisch niet-aangedane jongens met adrenoleukodystrofie toonde aan dat 80% van deze jongens al een verminderde bijnierschorsfunctie had ten tijde van de diagnose van adrenoleukodystrofie. De meest voorkomende symptomen van bijnierschorsinsufficiëntie zijn chronische of langdurige vermoeidheid, spierzwakte, verlies van eetlust, gewichtsverlies, buikpijn en onverklaarbaar braken. Andere mogelijke symptomen zijn misselijkheid, diarree, lage bloeddruk (die verder kan zakken bij opstaan, wat duizeligheid of flauwvallen kan veroorzaken), prikkelbaarheid en depressie, verlangen naar zout voedsel, lage bloedsuikerspiegel, hoofdpijn of zweten. Patiënten kunnen al dan niet toegenomen huidpigmentatie hebben, als gevolg van overmatige afgifte van het hormoon adrenocorticotropine (ACTH).
Myelopathie: bijna alle mannen ontwikkelen als volwassene schade aan het ruggenmerg (myelopathie). Dit ontstaat meestal op de leeftijd van 20-40 jaar. De symptomen worden veroorzaakt door schade aan het ruggenmerg en de perifere zenuwen van de benen. De klachten ontstaan doorgaans geleidelijk en nemen ook langzaam toe. Mede hierdoor wordt de diagnose adrenoleukodystrofie zelden gesteld in de eerste 3-5 jaar na ontstaan van de eerste klachten, tenzij er andere gevallen van adrenoleukodystrofie in dezelfde familie zijn vastgesteld. Patiënten ontwikkelen een langzaam progressieve loopstoornis als gevolg van zwakte, stijfheid en verminderd gevoel in de benen. Patiënten kunnen ook problemen met plassen en ontlasting (incontinentie) ontwikkelen. Ook deze symptomen zijn vaak progressief. Vaak zijn patiënten genoodzaakt om in de loop van de jaren met een hulpmiddel te lopen of een rolstoel te gebruiken.
Adrenomyeloneuropathie (AMN): De term AMN verwijst naar mannelijke patiënten met zowel een verminderde bijnierschorsfunctie als een myelopathie.
Cerebrale ALD: Ongeveer 40% van de jongens met adrenoleukodystrofie krijgt voor het 18de levensjaar een ontstekingsreactie in de hersenen (cerebrale ALD). Voor zover bekend ontstaat cerebrale ALD niet vóór de leeftijd van 3 jaar. In het verleden werd cerebrale ALD beschouwd als zeldzaam in de adolescentie (4-7%) en volwassenheid (2-5%). Systematisch onderzoek van een grote groep mannen met adrenoleukodystrofie die jaarlijkse MRI-scans krijgen wijst er echter op dat deze percentages hoger zijn. Helaas kunnen we niet voorspellen of en wanneer een patiënt cerebrale ALD zal ontwikkelen. Het ontstaan van cerebrale ALD wordt hoogstwaarschijnlijk bepaald door het samenspel van de mutatie in het ABCD1-gen en een combinatie van tot nu toe nog onbekende omgevingsfactoren en/of genetische factoren. Er zijn aanwijzingen dat ook een val (of ander trauma) op het hoofd in sommige gevallen cerebrale ALD kan uitlokken. De symptomen van cerebrale ALD zijn over het algemeen snel progressief. Bij jongens van basisschoolleeftijd zijn de eerste symptomen meestal gedragsproblemen en leerstoornissen, die zich uiten in een afname van de schoolprestaties. Deze vroege klachten worden vaak in eerste instantie toegeschreven aan andere stoornissen, zoals aandachtstekort-hyperactiviteitstoornis (ADHD), wat de diagnose van adrenoleukodystrofie kan vertragen. Bij volwassen patiënten zijn de eerste symptomen vaak psychiatrisch van aard en kunnen lijken op een depressie of psychose. Bij deze patiënten kan het jaren duren voordat de diagnose van adrenoleukodystrofie gesteld wordt; vooral als er geen familiegeschiedenis van adrenoleukodystrofie bekend is en wanneer andere klachten (van AMN) ontbreken. Naarmate de ziekte voortschrijdt, worden duidelijke neurologische klachten zichtbaar. Bijvoorbeeld stoornissen in gehoor- en gezichtsvermogen, zwakte van de ledematen, problemen met coördinatie en epileptische aanvallen. In dit stadium is de ziekteprogressie vaak zeer snel. Aangedane patiënten kunnen binnen een paar maanden het vermogen om taal te begrijpen verliezen en kunnen niet meer lopen. Uiteindelijk worden zij bedlegerig, blind, en zijn niet in staat om te spreken of te eten. De meeste cerebrale patiënten zijn 2 tot 4 jaar na het begin van de eerste symptomen in een zogenaamde ‘vegetatieve toestand’ of zijn overleden. Er zijn echter cerebrale patiënten beschreven die – indien goed verzorgd – jaren in deze ogenschijnlijke vegetatieve toestand kunnen blijven.
Vrouwen met ALD: Zoals bij veel X-gebonden ziekten, werd oorspronkelijk aangenomen dat vrouwen met het adrenoleukodystrofie gendefect vrij van symptomen blijven. Het is nu echter duidelijk dat deze veronderstelling onjuist is. Inmiddels is aangetoond dat meer dan 80% van de vrouwen met ALD op de leeftijd van 60 jaar symptomen en klachten te gevolge van adrenoleukodystrofie heeft ontwikkeld. Over het algemeen ontstaan de neurologische symptomen op een latere leeftijd dan bij mannen met een myelopathie; meestal tussen het 40ste en 50ste levensjaar. Het beloop van de ziekte is doorgaans ook langzamer dan bij aangedane mannen. In tegenstelling tot de aangedane mannen is incontinentie voor ontlasting een veel voorkomende klacht bij vrouwen met ALD. De ervaring leert dat de myelopathie bij vrouwen met ALD vaak verkeerd wordt gediagnosticeerd als multiple sclerose (MS). Vrouwen hebben een zeer kleine kans (minder dan 1%) op het ontwikkelen van een bijnierschorsinsufficiëntie of cerebrale ALD.
Diagnostiek
Adrenoleukodystrofie kan worden gediagnosticeerd door het aantonen van een verhoogde hoeveelheid verzadigde zeer langketen vetzuren in het bloed. Deze test is een betrouwbare en zeer nauwkeurige methode voor het diagnosticeren van adrenoleukodystrofie in mannen van alle leeftijden. Maar ongeveer 20% van de vrouwen met ALD heeft normale ZLKV in het bloed of in cellen. De test geeft in zo’n geval dus een “fout-negatief” resultaat. Een veelgebruikte manier om “fout-negatieve” resultaten te ondervangen is een combinatie van de ZLKV test met een DNA test. Deze combinatietest maakt het mogelijk om vrouwen met ALD te identificeren: normale resultaten voor zowel de biochemische als de genetische testen bevestigen dat een vrouw geen adrenoleukodystrofie heeft.
Hielprikscreening
Vroege diagnose van adrenoleukodystrofie is de sleutel tot het redden van levens. Screening van pasgeboren kinderen maakt het mogelijk om adrenoleukodystrofie patiënten tijdig op te sporen om zo het ontstaan van de bijnierschorsfunctie en/of cerebrale ALD in een heel vroeg stadium te diagnosticeren en behandeling te starten. Er is een speciale screeningstest voor pasgeborenen ontwikkeld. Deze meet de ZLKV niveaus (als C26:0-lysoPC) in hielprikbloed van pasgeborene. Op 30 december 2013 startte de staat New York met de hielprikscreening op adrenoleukodystrofie. In februari 2016 is adrenoleukodystrofie toegevoegd aan de “Recommended Uniform Screening Panel” (RUSP) in de Verenigde Staten. Sindsdien zijn in de VS meerdere staten begonnen met adrenoleukodystrofie neonatale screening. In 2015 is het hielprikprogramma in Nederland uitgebreid met 14 ziekten, waaronder adrenoleukodystrofie. De screening op adrenoleukodystrofie is echter nog niet begonnen. Op de pagina “Hielprik screening” kunt u gedetailleerde en actuele informatie over de hielprik voor adrenoleukodystrofie lezen.
Onderzoek
Over de hele wereld wordt uitgebreid onderzoek gedaan naar adrenoleukodystrofie. In 1993 werd het gen voor adrenoleukodystrofie geïdentificeerd door de gezamenlijke inspanningen van Drs. Patrick Aubourg en Jean-Louis Mandel in Frankrijk en Dr. Hugo Moser in de VS. Dit heeft deuren geopend voor verder onderzoek en het ontwikkelen van bijvoorbeeld gentherapie. Onderzoeksactiviteiten zijn gericht op vele aspecten van adrenoleukodystrofie. Zowel klinische vragen als meer fundamentele vragen, zoals: “Hoe veroorzaken de ZLKV schade aan de hersenen en het ruggenmerg?”; “Waarom ontwikkelt de ene patiënt cerebrale ALD terwijl een andere patiënt (deze patiënt kan zelfs een broer of familielid zijn) op latere leeftijd een myelopathie ontwikkelt?”
Behandeling
Er is nog geen genezende behandeling voor adrenoleukodystrofie.
Hormoonsuppletie: De meeste mannelijke adrenoleukodystrofie patiënten ontwikkelen bijnierschorsinsufficiëntie (tekort aan het stresshormoon cortisol). Bijnierschorsinsufficiëntie is vaak de eerste manifestatie van adrenoleukodystrofie. Bijnierschorsinsufficiëntie is te behandelen met hormoonsuppletie. Als de bijnierschorsinsufficiëntie echter niet op tijd wordt ontdekt, kan dit ernstige gevolgen hebben. Het ontstaan of behandelen van bijnierschorsinsufficiëntie heeft geen invloed op de kans op het ontwikkelen van neurologische symptomen.
Dieet: Omdat ZLKV schadelijk zijn voor zenuwcellen, het myeline, de bijnieren en de testis zijn er verschillende pogingen ondernomen om de bloedconcentraties van ZLKV te verlagen door aanpassingen in het dieet. Dieetaanpassingen gebaseerd op een beperking van de inname van ZLKV hebben geen effect op de ZLKV niveaus in bloed.
Lorenzo’s olie: Verzadigde ZLKV worden voornamelijk gemaakt vanuit kortere vetzuren. In cellen van adrenoleukodystrofie patiënten die in het laboratorium worden gekweekt kunnen we de verzadigde ZLKV verlagen door aan het kweekmedium mono-onverzadigde vetzuren toe te voegen. Deze bevinding kan worden verklaard omdat de enzymen die nodig zijn voor de aanmaak van verzadigde ZLKV dezelfde zijn als de enzymen die de mono-onverzadigde vetzuren maken. Hun affiniteit voor de mono-onverzadigde vetzuren is echter hoger. Deze ontdekking vormde de basis van een dieettherapie. Orale toediening van oliezuur in triglyceridevorm (GTO) en erucazuur in triglyceridevorm (GTE) normaliseerde de bloed ZLKV niveaus binnen 1 maand bij de meeste patiënten met adrenoleukodystrofie. De combinatie van GTO en GTE in een 4: 1-verhouding werd bekend als “Lorenzo’s oil”. Dit was een eerbetoon aan Lorenzo Odone, de eerste patiënt die met het mengsel werd behandeld. Vanwege het effect van Lorenzo’s olie op de ZLKV waarden in het bloed waren de verwachtingen hoog gespannen. Verschillende onderzoeken hebben echter aangetoond dat de olie schade aan de bijnieren of het zenuwstelsel niet remt. Meer informatie is te lezen (in het Engels) op de pagina (Lorenzo’s oil).
Lovastatine: In 1998 werd aangetoond dat het cholesterolverlagende medicijn lovastatine een effect had op ZLKV. Deze bevinding kon echter niet door anderen worden gereproduceerd. Resultaten van andere onderzoekers lieten zien dat cholesterolverlagende medicijnen geen effect hadden op ZLKV niveaus in de hersenen en bijnieren van adrenoleukodystrofie muizen. In aantal gevallen resulteerde behandeling zelfs in een toename van ZLKV in deze weefsels. Om duidelijkheid te verschaffen in deze tegenstrijdige resultaten is er een gerandomiseerde, dubbelblinde, placebo-gecontroleerde klinische studie uitgevoerd in het Academisch Medisch Centrum in Amsterdam om het effect van lovastatine als een ZLKV-verlagende therapie voor adrenoleukodystrofie te testen. De resultaten en conclusies tonen duidelijk aan dat behandeling met lovastatine inderdaad resulteert in een kleine afname van ZLKV in het bloed. Echter op cellulair niveau gebeurde er niets: de ZLKV niveaus in rode- en witte bloedcellen bleven onveranderd. Meer details (in het Engels) kunt u lezen op de pagina (Lovastatin).
Bezafibraat: Omdat ZLKV vooral door het lichaam zelf gemaakt worden zou het mogelijk moeten zijn dit proces af te remmen. In een onderzoek, waarin stoffen getest werden die mogelijk een effect op het ZLKV metabolisme kunnen hebben, werd bezafibraat gevonden als een medicijn dat ZLKV kan verlagen in cellen van adrenoleukodystrofie patiënten. Bezafibraat is een geneesmiddel dat wordt gebruikt voor de behandeling van hyperlipidemie. Experimenten in fibroblasten toonden aan dat bezafibraat de ZLKV niveaus verlaagde door de activiteit van het ZLKV-specifieke elongase ELOVL1 (het enzym dat verantwoordelijk is voor de aanmaak van ZLKV) direct te remmen. Om het effect van bezafibraat op ZLKV-accumulatie in bloedcellen van AMN patiënten te evalueren werd er een open-label pilotstudie uitgevoerd in het Academisch Medisch Centrum in Amsterdam. Helaas kon bezafibraat de ZLKV waarden in bloedcellen van ALD patiënten niet verlagen. Hoogstwaarschijnlijk was dit te wijten aan een te lage dosering. De dosering die is gebruikt in de klinische studie is de maximale toegestane dosering voor patiënten. De effectieve dosis in fibroblasten was ongeveer 20x hoger.
Beenmergtransplantatie: Een beenmerg- of hematopoëtische stamceltransplantatie kan de progressie van cerebrale ALD in jongens en adolescenten stoppen. Een belangrijke voorwaarde is dat de transplantatie in een zeer vroeg stadium van de ziekte wordt uitgevoerd. Het is nog steeds niet helemaal duidelijk waarom de stamceltransplantatie zo effectief is. De meest geaccepteerde verklaring is dat de stamceltransplantatie ervoor zorgt dat de niet goed functionerende microgliacellen (dit zijn cellen die in de hersenen een belangrijke rol spelen bij het tegengaan van ontstekingsreacties) vervangen worden door normale microgliacellen die afkomstig zijn van de stamcellen van de donor. Meer details (in het Engels) kunt u lezen op de pagina (Hematopoietic stem cell transplantation).
Gentherapie: Naar verwachting zal, in de niet al te verre toekomst, een transplantatie van in het laboratorium genetisch gecorrigeerde hematopoëtische stamcellen afkomstig van de patiënt zelf mogelijk worden. Deze methode is gebaseerd op de zeer bemoedigende resultaten van een onderzoek met 2 adrenoleukodystrofie patiënten. Meer details en informatie (in het Engels) over de studie in een grotere groep patiënten kunt u lezen op de pagina (Gene Therapy for ALD).
ALD uitgelegd in 10 minuten
Deze video (in het Engels) geeft een mooi en beeldend overzicht van adrenoleukodystrofie.
Geproduceerd door Youreka Science in samenwerking met ALD Connect, Inc.
Op de pagina (ALD Connect Educational Videos & Webinars page) vindt u meer videos en webinars (in het Engels).
Zeer langketen vetzuren
Wat is, biochemisch gezien, het probleem bij ALD/AMN?
Patiënten met adrenoleukodystrofie (ALD) hebben een verhoogde hoeveelheid (stapeling) van verzadigde zeer langketen vetzuren (ZLKV) in cellen, weefsels en organen. Deze stapeling is het grootst in de hersenen, ruggenmerg en bijnieren. Een te hoge concentratie ZLKV is schadelijk voor cellen. In het ruggenmerg en hersenen veroorzaakt dit neurologische problemen en in bijnieren resulteert de schade in het ontstaan van bijnierschorsinsufficiëntie. ZLKV accumuleren ook in het bloed. Dit maakt het mogelijk om ALD te diagnosticeren met behulp van een bloedtest.
Normale plasma ZLKV niveaus in een draagster voor ALD. Wat betekent dit?
De ZLKV bepaling in bloed is nog steeds de meest gebruikte diagnostische test voor ALD. De test meet 3 vetzuren: C22:0, C24:0 en C26:0. Deze test is een betrouwbare en zeer nauwkeurige methode voor het diagnosticeren van ALD in mannen van alle leeftijden. Maar ongeveer 20% van de vrouwen met ALD heeft normale ZLKV in het bloed of in cellen. De test geeft in zo’n geval dus een “fout-negatief” resultaat.
Voor vrouwen met de verdenking ALD geldt dat een normaal ZLKV niveau in bloed de diagnose ALD niet uitsluit. Een veelgebruikte manier om “fout-negatieve” resultaten te ondervangen is een combinatie van de ZLKV test met een DNA test. Deze combinatietest maakt het mogelijk om vrouwen met ALD te identificeren: normale resultaten voor zowel de biochemische als de genetische testen bevestigen dat een vrouw geen ALD heeft.
Wat zijn vetzuren?
Om de chemische structuur van vetzuren uit te kunnen leggen is een kleine basiskennis van scheikunde nodig.
De meeste chemische stoffen die we kennen, zoals bijvoorbeeld zout en water, zijn opgebouwd uit verschillende bouwstenen ook wel “atomen of elementen” genoemd. Zout is opgebouwd uit de atomen natrium (Na) en chloor (Cl). De chemische structuur van zout is erg simpel: één zout atoom is gekoppeld aan één chloor atoom. De aantrekkingskracht die de atomen in een molecuul bijeen houdt noemen we de chemische binding. Natrium kan slechts één ander atoom binden; hetzelfde geldt voor chloor. De chemische structuur van het molecuul zout kunnen we dan ook beschrijven als Na-Cl. De lijn tussen het Na en het Cl atoom geeft de chemische binding aan.
Water is iets gecompliceerder. Het is opgebouwd uit de atomen waterstof (H) en zuurstof (O). Waterstof kan, net als Na en Cl, een chemische binding aangaan met maar één ander atoom. Zuurstof daarentegen kan twee chemische bindingen aangaan. In een molecuul water binden twee atomen H aan één atoom O. De structuur van water kunnen we dus ook beschrijven als H-O-H.
Het gegeven dat atomen bindingen aan kunnen gaan met andere atomen heeft geresulteerd in het voorkomen van een bijna oneindige hoeveelheid verschillende chemische verbindingen. Wij en alles in onze omgeving bestaan uit scheikundige verbindingen.
Vetzuren zijn opgebouwd uit de elementen H, O, en koolstof (C). Koolstof kan vier chemische bindingen aangaan; dit maakt koolstof tot een zeer beweeglijk atoom.
De (vet) staart van een vetzuur is een keten van aan elkaar gebonden koolstofatomen waarbij ieder koolstofatoom ook met een aantal H atomen gebonden is.

Het “zuur” gedeelte (de zuurgroep) van een vetzuur bevat één C, twee O’s en één H. In de zuurgroep zien we een dubbele lijn tussen de C en de twee O’s. Dit noemen we een “dubbele binding”.

Voorbeelden van vetzuren
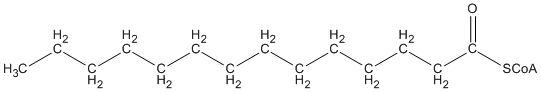
Azijnzuur, een kort-keten vetzuur; C2:0

Palmitinezuur of palmitaat is een lang-keten vetzuur; C16:0

Twee zeer lang-keten vetzuren (ZLKV) zijn tetracosanoaat (lignocerinezuur); C24:0

en hexacosanoaat; C26:0

In bovenstaande voorbeelden ziet u ook dat er een handige manier is om de ingewikkelde namen af te korten. Dit gaat als volgt: het getal dat weergegeven staat na de “C” geeft aan uit hoeveel koolstoffen de vetstaart bestaat, bijv. C2, C16, C24, C26, etc. Het getal na de dubbele punt geeft het aantal dubbele bindingen in de koolstofketen aan (de mate van verzadiging). Alle vetzuren hierboven weergegeven hebben geen dubbele bindingen tussen de koolstoffen en dus schrijven we bij allemaal “:0”.
Vetzuren – Wat heb je er aan? Waar komen ze vandaan en wat doet ons lichaam ermee?
Het lichaam kan niet bestaan zonder vetzuren. Ze komen voor in complexe chemische verbindingen zoals triglyceriden, fosfolipiden, sfingolipiden, glycolipiden, etc. Triglyceriden dienen vooral voor de vetopslag. Fosfolipiden vormen een belangrijk bestanddeel van de celmembraan en de membranen van de organellen in de cel. Sfingolipiden and glycolipiden zijn complexe verbindingen die vooral voorkomen in de hersenen en de zenuwcellen; gangliosiden en myeline behoren tot deze categorie. Het komt er eigenlijk op neer dat vetzuren essentieel zijn voor zeer veel normale lichaamsfuncties. Zonder vetzuren is er geen leven mogelijk.
Vetzuren komen voor in ons dagelijks dieet. Met name “vet” voedsel zoals gefrituurde producten en oliën, maar ook noten en zaden bevatten grote hoeveelheden vetten. Vlees, zelfs mager vlees, bevat een flinke hoeveelheid vetzuren. Daarentegen vinden we weinig vet in groente, fruit en zetmeelrijk voedsel (bijv. pasta en brood).
Wat gebeurt er met de vetzuren uit het dieet? Vetzuren en andere nutriënten uit ons voedsel komen in de maag waar de spijsvertering start. Tijdens dit proces wordt het voedsel afgebroken tot componenten die door het lichaam kunnen worden opgenomen, zoals koolhydraten (suiker en zetmeel), eiwitten en lipiden (vetzuren en cholesterol). Deze voedselcomponenten kunnen vervolgens door de cellen in de wand van de dunne darm opgenomen worden. De nutriënten passeren de darmwandcellen en worden opgenomen in kleine bloedvaten van de dunne darm. Voordat dit bloed de rest van het lichaam bereikt, passeert het eerst de lever. De lever zou je kunnen zien als een verwerkingsbedrijf voor nutriënten. In de cellen van de lever worden de nutriënten gemetaboliseerd; dit betekent dat ze 1) afgebroken worden voor de productie van warmte of energie, of 2) omgezet worden in bouwstoffen die elders in het lichaam nodig zijn. Een teveel aan nutriënten wordt omgezet naar een vorm die opgeslagen kan worden zoals glycogeen (suikers) en triglyceriden (vetzuren).
Vaak komt het voor dat er meer vetzuren aanwezig zijn dan het lichaam nodig heeft. Dit kan komen doordat we of teveel gegeten hebben of omdat het lichaam zelf teveel aanmaakt. Hoe komen we van het teveel aan vetzuren af? Vetzuren worden afgebroken of “geoxideerd” waarbij energie of warmte voor het lichaam vrijkomt. Vetzuren zijn de belangrijkste leveranciers van energie voor het lichaam in tijden van voedselschaarste. Er bestaat een delicate balans tussen het hebben van net genoeg of teveel aan vetzuren. Normaal gesproken staat het lichaam zeer fijntjes afgesteld om deze balans in evenwicht te kunnen houden. Het doorslaan van de balans naar één kant resulteert meestal in het ontstaan van ziekte.
ZLKV: Wat weten we van ze?
1) Zijn ze lichaamseigen?
– JA!
2) Wat is hun functie?
– Ze maken onderdeel uit van membranen, waaronder het myeline, de “isolatielaag” rondom zenuwen.
3) Waar komen ze vandaan?
– Dieet en d.m.v. verlenging van kortere vetzuren in het lichaam
4) Wat kan de verhoogde ZLKV niveaus in ALD veroorzaken?
– Het lichaam maakt te veel
– Het lichaam kan het overschot aan ZLKV niet afbreken
5) Welke van deze mogelijkheden is de juiste?
Studies uitgevoerd in cellen van patiënten en controle personen hebben aangetoond dat het proces dat normaal gesproken de ZLKV afbreekt tot kortere vetzuren niet goed functioneert in ALD.
Hoe breekt het lichaam vetzuren af?
Vetzuren worden afgebroken (korter gemaakt) in een serie opeenvolgende chemische reacties waarbij er iedere keer twee koolstoffen verwijderd worden:
Vetzuuroxidatie

Een vetzuur is chemisch gezien niet erg reactief.
Het enzym “fatty acyl-CoA synthetase” voegt een “coenzym A” toe aan het vetzuur.

(“geactiveerd” vetzuur)
Het enzym “acyl-CoA oxidase” introduceert een “dubbele binding”.

Het enzym “enoyl-CoA hydratase” gebruikt een water molecuul om de dubbele binding te verzadigen en een hydroxyl (O-H) groep te introduceren.

De “O-H” wordt geoxideerd tot een “=O” door het enzym “hydroxyacyl-CoA dehydrogenase”.

Het C16 vetzuur wordt door het enzym “thiolase” gesplitst in een C14 vetzuur en een C2 vetzuur.
 +
+ 
Het C14 vetzuur kan vervolgens weer opnieuw de cyclus beginnen waarbij er een C12 vetzuur ontstaat. De hierdoor vrijgekomen C2 vetzuren worden gebruikt voor de productie van energie.
Hoe werken enzymen?

Een enzym maakt een chemische reactie in of buiten een cel mogelijk of versnelt deze zonder daarbij zelf verbruikt te worden of van samenstelling te veranderen. Enzymen hebben specifieke bindingsplaatsen voor de chemische stoffen (substraten) waarmee ze een interactie aangaan. Het figuur toont de eerste reactie van de vetzuuroxidatie. Het enzym heeft één bindingsplaats voor een vetzuur en een tweede voor het coenzym A (CoA). Deze bindingsplaatsen zijn zeer specifiek en je kunt ze vergelijken als een sleutel en een slot. Doordat het enzym de twee substraten dicht bij elkaar brengt, kunnen ze elkaar binden. Hierdoor ontstaat er een nieuwe chemische verbinding, een geactiveerd vetzuur (acyl-CoA vetzuur). Na de reactie laat het enzym het geactiveerde vetzuur los, waarna het enzym weer een volgend vetzuur en CoA kan binden. Zonder de aanwezigheid van het enzym is de kans dat een vetzuur een CoA bindt nihil.
Wat is het “ALD proteïne” en wat doet het?

Een cel bevat een aantal kleinere compartimenten die organellen worden genoemd. Het creëren van afgesloten ruimtes heeft de cel de mogelijkheid gegeven om enzymen die moeten samenwerken bij elkaar te houden in hetzelfde compartiment. Vetzuurafbraak (vetzuuroxidatie) vindt plaats in twee verschillende organellen – het mitochondrium en het peroxisoom. De enzymen die betrokken zijn bij de vetzuurafbraak in mitochondria zijn anders dan die in de peroxisomen. Onderzoek uitgevoerd in het Kennedy Krieger Instituut in de jaren 80 door Dr. Inderjit Singh en Dr. Hugo Moser heeft aangetoond dat de afbraak van ZLKV uitsluitend uitgevoerd wordt in de peroxisomen.
In 1993, is door de onderzoeksgroep van Dr. Patrick Aubourg en Dr. Jean-Louis Mandel in Parijs aangetoond dat het door het ALD gen geproduceerde ALD proteïne (ALDP) in cellen gelokaliseerd is in het peroxisoom. Het ALDP behoort tot een specifieke familie van transporteiwitten (ABC transporters). ABC transporteiwitten verbruiken energie voor het verplaatsen van een molecuul (het substraat) van de ene naar de andere zijde van het membraan (dus van buiten de cel naar binnen of andersom, of een compartiment (binnen de cel) in of uit).
Het ALDP fungeert als een soort poort in het membraan van het peroxisoom. ZLKV gaan door de poort het peroxisoom binnen waar ze afgebroken kunnen worden voor de productie van energie.
De meest voor de hand liggende verklaring voor wat er mis gaat bij de ziekte ALD is dat een mutatie in het ALDP ervoor zorgt dat het ALD proteïne zijn transport functie niet kan vervullen. Het gevolg is dat de vetzuren in het cytoplasma die wachten op transport naar het peroxisoom toe niet weg kunnen en stapelen.
Hielprik Screening
Authors: Rachel Salzman, Rinse Barendsen en Stephan Kemp
Introductie
Adrenoleukodystrofie (ALD) is een erfelijke metabole ziekte waarbij zeer-lange-keten vetzuren niet door het lichaam kunnen worden afgebroken. Ophoping van deze vetten kan leiden tot schade aan de hersenen, bijnieren en het ruggenmerg. De klachten en de ernst van deze klachten kunnen wisselen. Ongeveer de helft van de jongens met ALD krijgt voor het tiende levensjaar schade aan de bijnieren. Bij 1 op de 3 jongens met ALD ontstaat tussen het derde en tiende levensjaar een ernstige ontsteking in de hersenen (cerebrale ALD).
Kinderen die worden geboren met ALD hebben neurologisch geen afwijkingen bij de geboorte. Een vroege diagnose van jongens met ALD kan leiden tot levensreddende interventies.
Deze levensreddende interventies bestaan uit twee delen:
- Op tijd starten met suppletie van bijnierhormonen als er sprake is van bijnierschorsinsufficiëntie.
- Op tijd behandelen van de ontstekingsreactie in de hersenen (cerebrale ALD) met een allogene beenmerg- of hematopoëtische stamceltransplantatie.
Een stamceltransplantatie kan de vaak fatale progressie van cerebrale ALD stoppen, mits de procedure in een zeer vroeg stadium van de ziekte wordt uitgevoerd. Helaas is er maar een krap therapeutisch venster waarin deze behandeling effectief is. Door het onvoorspelbare beloop van de ziekte wordt de diagnose ALD vaak te laat gesteld om nog te kunnen behandelen. Het screenen van pasgeboren op ALD door middel van de hielprik maakt een vroege diagnose van ALD mogelijk. Hierdoor wordt een tijdige behandeling mogelijk gemaakt.
In februari 2016 is in de Verenigde Staten ALD toegevoegd aan het “Recommended Uniform Screening Panel (RUSP)”. De RUSP is de federale lijst waarin alle genetische ziektes die worden aanbevolen voor neonatale hielprikscreening staan vermeld. De staat New York is op 30 december 2013 begonnen met de screening op ALD bij pasgeborenen. Sindsdien zijn ook andere staten gestart met de neonatale screening op ALD (Figuur 1). De verwachting is dat de overige staten later zullen volgen zodra hiervoor de financiële middelen, follow-up protocollen en logistiek beschikbaar zijn.

Figuur 1: Kaart met de staten in de VS waar pasgeborenen gescreend worden op ALD.
Nederland

In Nederland heeft de minister van Volksgezondheid opdracht gegeven om een pilotstudie te verrichten naar het toevoegen van ALD aan de landelijke neonatale hielprikscreening. De voorbereidingen voor de pilot zijn gestart in december 2018. Dit betrof eerst nog veel voorbereidend werk. Vanaf 1 januari 2021 t/m 31 december 2021 is het hielprikbloed in vier provincies (Noord-Holland, Flevoland, Utrecht en Gelderland) getest op ALD. Het doel van deze pilot is om te onderzoeken hoe de screening van ALD het beste in de huidige neonatale hielprikscreening geïmplementeerd kan worden. De naam van de pilotstudie is SCAN-studie (SCreening op ALD in Nederland). Meer informatie is te vinden op www.scanstudie.nl.
Naar aanleiding van de succesvolle pilotstudie heeft de staatssecretaris van Ministerie van Volksgezondheid, Welzijn en Sport besloten om ALD vanaf 1 oktober 2023 toe te voegen aan de hielprikscreening (Zie ook: Stofwisselingsziekte ALD wordt onderdeel van de hielprikscreening).
Criteria voor opname van een ziekte in de hielprikscreening
Er is brede internationale consensus over de criteria waar een ziekte aan moet voldoen om in aanmerking te komen voor opname in de landelijke neonatale hielprikscreening:
- Vroegtijdige diagnose moet voor de pasgeborene direct voordelig zijn. Er moet sprake zijn van substantiële gezondheidswinst door de vroege diagnose en het kunnen starten van behandeling.
- De screeningstest moet van zeer goede kwaliteit zijn. De test moet een hoge specificiteit en sensitiviteit hebben. Dit betekent dat er een zeer lage kans mag zijn op zowel fout-positieve als fout-negatieve resultaten.
 Geschiedenis
Geschiedenis
In 2004 stelde dr. Hugo Moser in een bijeenkomst van de Amerikaanse National Advisory Committee for Newborn Screening voor om ALD toe te voegen aan de RUSP. Het probleem was echter dat er op dat moment nog geen test bestond voor screening op ALD bij pasgeborenen. Om dit probleem op te lossen organiseerde dr. Hugo Moser de benodigde financiële middelen en startte hij een onderzoek in het Kennedy Krieger Institute (Baltimore, MD), met als doel een geschikte biomarker voor ALD te vinden. In 2006 identificeerde het team C26:0-lysofosfatidylcholine (C26:0-LPC) in postnatale veneuze bloedspots van mannelijke pasgeborenen met ALD als bruikbare biomarker (Hubbard et al. 2006). In de daaropvolgende jaren bleven wetenschappers de analyse verbeteren (Hubbard et al. 2009; Theda et al. 2014). Samen met onderzoekers van de Mayo Clinic (Rochester, Minnesota) werd vervolgens een high-throughput-methode ontwikkeld voor de analyse van C26:0-LPC (Haynes and De Jesús 2012; Turgeon et al. 2015). In 2013 werd deze methode gevalideerd met behulp van 100.000 anonieme bloedspots.
 Aidan’s Law
Aidan’s Law
De familie Seeger heeft ervoor gezorgd dat in de staat New York gestart werd met het screenen van pasgeborenen op ALD. Dit deden zij ter nagedachtenis aan hun zoon, Aidan, die cerebrale ALD had. Door een te laat gestelde diagnose is Aidan in 2012 overleden aan de gevolgen van cerebrale ALD. Om te voorkomen dat meer families dit leed moesten doormaken is de familie Seeger een uitgebreide lobby gestart. In april 2012 werd in de staat New York een wetsvoorstel ingediend voor het screenen van pasgeborenen op ALD. Deze wet, “Aidan’s Law”, werd in februari 2013 aangenomen. In december 2013 kon dankzij “Aidan’s Law” ALD worden toegevoegd aan de hielprikscreening in de staat New York.
New York State
Tijdens de eerste drie jaar dat in New York State werd gescreend op ALD, zijn meer dan 700.000 pasgeboren kinderen gescreend. Dankzij deze screening op ALD konden 45 baby’s met ALD worden geïdentificeerd: 22 jongens en 23 meisjes. Op basis van deze aantallen kon de incidentie van ALD bij de geboorte worden bepaald op 1 op 15.000.
Wanneer een pasgeborene met ALD wordt geïdentificeerd, wordt de behandelend arts van het gezin op de hoogte gebracht. Het gezin wordt door de behandelend arts doorverwezen naar een klinisch geneticus voor bevestiging van de diagnose. Hierbij wordt ook genetische counseling aangeboden. Een andere dienst die ouders wordt aangeboden is uitgebreide familiescreening naar een risico op ALD.
Het screeningsalgoritme
In de Verenigde Staten wordt in de meeste staten de diagnose ALD gesteld met behulp van een drieledig screeningsalgoritme (Figuur. 2). De eerste test is een high-throughput standaard MS/MS-analyse van C26:0-LPC. Monsters met een verhoogde C26:0-LPC-concentratie gaan vervolgens door naar de tweede stap. De tweede stap bestaat uit analyse van C26:0-LPC door middel van HPLC-MS/MS. Deze test is specifieker, maar duurt ook iets langer dan de eerste stap. De monsters die ook in de tweede stap een verhoogde C26:0-LPC hebben, gaan door naar stap 3. Stap 3 is het stellen van de diagnose ALD door middel van DNA-analyse. Op het DNA wordt specifiek gezocht naar een mutatie in het ABCD1-gen.
 Figuur 2: Het drieledige screeningsalgoritme gebruikt in de Verenigde Staten.
Figuur 2: Het drieledige screeningsalgoritme gebruikt in de Verenigde Staten.
Uitdagingen
In verschillende landen zijn er aanzienlijke uitdagingen en voortdurende ethische discussies met betrekking tot de opname van ALD in de neonatale hielprikscreening voor pasgeborenen. De belangrijkste uitdagingen:
- Het eerste criterium voor inclusie in de nationale hielprikscreening schrijft voor dat vroege diagnose direct voordelig moet zijn voor de pasgeborene. Dit kan bij ALD reden geven voor ethische bezwaren. Bij ALD ontwikkelt ongeveer een derde van de jongens cerebrale ALD tussen de leeftijd van 3 en 18 jaar. De resterende twee derde van de mannen zal adrenomyeloneuropathie (AMN) ontwikkelen op de volwassen leeftijd. AMN wordt gekenmerkt door spasticiteit van de ledematen, loopstoornissen en incontinentie en wordt symptomatisch behandeld. De afwezigheid van biochemische markers of andere biologische hulpmiddelen maakt het voorspellen van gezondheidsresultaten voor individuen moeilijk en kan daarom het risico op onnodige medische interventies verhogen..
- Het screenen van pasgeborenen op ALD identificeert ook meisjes met een mutatie op het ABCD1-gen. Vrouwen met ALD hebben een << 1% kans op het ontwikkelen van bijnierschorsinsufficiëntie of cerebrale ALD. Er is dus geen direct gezondheidsvoordeel bij een vroege diagnose voor een pasgeboren meisje met ALD. Ongeveer 80% van de vrouwen met ALD zal op de leeftijd van 60 jaar een myelopathie ontwikkelen. Voor deze vorm is momenteel nog geen behandeling beschikbaar.
- In verschillende landen is er een groeiend debat binnen de wetenschappelijke gemeenschap en tussen patiëntenorganisaties over de opname van bepaalde onbehandelbare aandoeningen in landelijke hielprikprogramma’s. Zoals eerder vermeld, moet een vroege diagnose uiteindelijk resulteren in een direct gezondheidsvoordeel voor de pasgeborene zelf. Dit criterium is wellicht niet evident aanwezig in het geval van een ziekte die voor vrouwen niet behandelbaar is.
- Het kan voorkomen dat bij het screenen op een bepaalde ziekte, ziekten buiten het kader van de beoogde test worden gevonden. De huidige screeningstest op ALD kan ook niet-behandelbare ziektes detecteren die gepaard gaat met verhoogde niveaus van C26:0-LPC (Fig. 2). Deze omvatten: de Zellweger-spectrumstoornissen, de peroxisomale vetzuuroxidatiestoornissen veroorzaakt door een defect in het peroxisomale acyl-CoA-oxidase 1 (ACOX1) danwel het multifunctionele eiwit (HSD17B4), het “gecombineerde ABCD1 DXS1357E-deletiesyndroom” (CADDS), Acyl-CoA-Binding Domain-Containing Protein 5 (ACBD5)-deficiëntie en het Aicardi Goutières-syndroom.
- Buiten de eerdere genoemde voordelen, zoals het tijdig kunnen behandelen van bepaalde vormen van ALD, zijn nog andere voordelen te bedenken. Enkele voordelen zijn specifiek voor de pasgeborene. Een voorbeeld hiervan is het snellere diagnostische proces. Vanwege de aspecifieke klachten van ALD kan het buiten de hielprikscreening om vaak lang duren voor de diagnose ALD wordt gesteld.
- De meeste voordelen zijn echter vooral voor het gezin. Bij de diagnose ALD ontstaat namelijk de kans om gebruik te maken van uitgebreide familiescreening om extra familieleden die risico lopen te identificeren. Ook kunnen aanpassingen gedaan worden aan het familieleven om de gevolgen van de ziekte op te vangen. Daarnaast kunnen ouders profiteren van screening op een aandoening waarvoor geen effectieve behandeling bestaat, omdat deze kennis ouders informatie verschaft die ze kunnen gebruiken om in de toekomst keuzes te maken omtrent het krijgen van kinderen.
- Er zijn echter ook duidelijke nadelen. Het belangrijkste nadeel is dat de diagnose van een onbehandelbare ziekte een negatieve invloed kan hebben op, of stigma kan zijn voor het leven van de pasgeborene
Als gevolg van de toevoeging aan het RUSP wordt verwacht dat de komende jaren in een groeiend aantal staten in de VS en ook internationaal gestart zal worden met het screenen van pasgeborenen op ALD. Deze maatregelen zullen de klinische uitkomst van honderden pasgeborenen met ALD, hun biologische verwanten en hun geliefden aanzienlijk verbeteren.
Conclusie
De opname van ALD in de neonatale hielprikscreening markeert een belangrijke mijlpaal in de vroege detectie en behandeling van deze complexe aandoening. De vroege diagnose maakt tijdige interventies mogelijk, wat levensreddend kan zijn voor jongens met ALD. Daarnaast biedt het gezinnen de mogelijkheid om zich voor te bereiden op de toekomst en geïnformeerde beslissingen te nemen. Naarmate meer landen ALD-screening implementeren, zal onze kennis over de ziekte en de effectiviteit van vroege interventies alleen maar toenemen. Dit zal uiteindelijk leiden tot verbeterde zorg en levenskwaliteit voor individuen met ALD en hun families.
Vrouwen met ALD
Auteurs | Drs. Irene Huffnagel (arts-onderzoeker) | Dr. Stephan Kemp (onderzoeker) | Dr. Marc Engelen ((kinder)neuroloog)
ALD werd jarenlang beschouwd als een ziekte die alleen jongens en mannen treft. Onderzoek in het AMC onder een grote groep “draagsters” heeft laten zien dat dit beeld niet klopt. Vrouwen met een defect ABCD1-gen ontwikkelen ook neurologische klachten door ruggenmergschade (myelopathie). Maar, bijnierschorsinsufficiëntie en cerebrale ALD zijn heel zeldzaam bij vrouwen met ALD.
Deze pagina behandelt de klachten die vrouwen met ALD kunnen ontwikkelen en bespreekt wat eraan gedaan kan worden.
Introductie
Over het algemeen geldt dat X-chromosoom gebonden ziekten vooral mannen treffen, aangezien mannen (‘XY’) maar één X chromosoom hebben. Inmiddels is door onderzoek duidelijk geworden dat deze gedachte niet altijd opgaat. Voor een aantal X-gebonden ziekten geldt dat aangedane vrouwen (‘XX’) ook klachten kunnen ontwikkelen. ALD is hiervan een voorbeeld. Vrouwen met ALD zijn niet alleen draagsters van het defecte ABCD1-gen, maar ook patiënten. Omdat dit vaak niet bekend is, worden klachten regelmatig toegeschreven aan andere oorzaken. Hierdoor kan het langer duren voordat adequate behandeling van klachten gestart wordt. Zo zijn er een aantal gevallen bekend van vrouwen die operaties ondergingen (plaatsing van een heupprothese, operatie van de cervicale wervelkolom), sommige vrouwen zelfs meerdere malen, voor wat later symptomen van een ruggenmergziekte ten gevolge van ALD bleken te zijn. Naast onnodige behandelingen kan het niet herkennen van ALD bij een vrouw voorkomen dat de vrouw adequate genetische voorlichting krijgt over de erfelijkheid en vertraagt het de diagnose en behandeling van familieleden.
Het verhaal van een “draagster”
Mevrouw H. was 38 jaar, geboren in Nederland, maar woonde met haar familie in de Verenigde Staten. Ze had gemerkt dat het steeds moeilijker werd om haar evenwicht te bewaren. Ook begonnen haar benen een beetje stijf aan te voelen. Ze had al een aantal jaren problemen met plassen, maar ze dacht dat de bevalling van haar twee kinderen dit had veroorzaakt. Een van haar neven, waarvan aanvankelijk gedacht werd dat hij een hersentumor had, overleed toen hij 13 jaar oud was. Een andere neef, die toenemende loopproblemen had, werd gediagnosticeerd met multiple sclerose (MS). Nadat ze in 1985 een artikel over ALD had gelezen in een populair tijdschrift, herkende ze haar symptomen en die van haar neven. Zij besloot contact op te nemen met de arts die in het artikel genoemd werd: Dr. Hugo Moser van het Kennedy Krieger Institute in Baltimore, Verenigde Staten. Er werd een bloedmonster genomen en de analyse liet een verhoging zien van de zeer langketen vetzuren (ZLKV), waarmee de diagnose ALD werd bevestigd.
Jaren later bezocht mevrouw H. op 55 jarige leeftijd het Academisch Medisch Centrum in Amsterdam. De klachten van haar benen waren langzaam toegenomen. Binnenshuis kon ze zich met ondersteuning voortbewegen en buitenshuis gebruikte ze een scootmobiel. Opvallend was dat ondanks dat sommige van haar familieleden in de medische literatuur werden vermeld, er ook velen waren die nog niet waren onderzocht op ALD. Een aantal familieleden had er bewust voor gekozen om zichzelf niet te laten testen. Maar er waren ook familieleden met wie de mogelijkheid van testen op ALD nog nooit was besproken. Bij het lichamelijk onderzoek waren er tekenen van ruggenmergschade: een verhoogde spanning in de beenspieren, zwakte en afwijkende reflexen. Een MRI onderzoek liet geen afwijkingen zien aan de hersenen of het ruggenmerg. Mevrouw H. werd doorverwezen naar een revalidatiearts en een fysiotherapeut. De familie werd genetische voorlichting en een bloedtest voor ALD aangeboden. Terugkijkend werd vastgesteld dat haar neefje van 13 was overleden aan de gevolgen van cerebrale ALD. Er werden ook een aantal neven gediagnosticeerd met AMN. Samen met haar man, de voorzitter van de voormalige Nederlandse ALD patiëntenorganisatie (deze is opgenomen in de VKS), heeft mevrouw H. zich sindsdien vele jaren ingezet om nieuw gediagnosticeerde families en patiënten te helpen en ondersteunen.
De klachten die vrouwen met ALD kunnen ontwikkelen
Ondanks dat het sinds de jaren tachtig bekend is dat vrouwen met ALD klachten kunnen ontwikkelen is er pas in 2014 een groot onderzoek gepubliceerd waarin deze klachten systematisch in kaart gebracht zijn. Dit onderzoek werd uitgevoerd in het AMC en er deden 46 vrouwen aan mee. Het onderzoek liet zien dat hoe ouder de vrouwen waren hoe meer vrouwen er klachten hadden van ruggenmergschade door ALD (Figuur 1). Op de leeftijd van 60 jaar had zelfs meer dan 80% van de vrouwen neurologische klachten.
 Figuur 1: Correlatie tussen de symptomatische status en leeftijd in een cohort van 46 vrouwen met ALD. De groene balken geven het percentage vrouwen in elke leeftijdsgroep aan die klinische symptomen hebben ontwikkeld die veroorzaakt worden door ALD. De zwarte stippen tonen elk individu in het cohort aan. Zij zijn aangemerkt als symptomatisch (klachten en tekenen van ruggenmergschade) of niet-symptomatisch.
Figuur 1: Correlatie tussen de symptomatische status en leeftijd in een cohort van 46 vrouwen met ALD. De groene balken geven het percentage vrouwen in elke leeftijdsgroep aan die klinische symptomen hebben ontwikkeld die veroorzaakt worden door ALD. De zwarte stippen tonen elk individu in het cohort aan. Zij zijn aangemerkt als symptomatisch (klachten en tekenen van ruggenmergschade) of niet-symptomatisch.
Het ontwikkelen van klachten op de kinderleeftijd is bij vrouwen extreem uitzonderlijk. In tegenstelling tot mannen met ALD, hebben vrouwen met ALD een zeer kleine kans (minder dan 1%) op het ontwikkelen van een bijnierschorsinsufficiëntie of cerebrale ALD. De klachten die vrouwen met ALD kunnen ontwikkelen komen door schade aan het ruggenmerg en de zenuwen in de benen. De ziekte wordt langzaam erger. Over een periode van tientallen jaren kunnen vrouwen klachten ontwikkelen van zwakte en/of spasticiteit in de benen, een verstoord gevoel in de benen en/of incontinentie voor urine en/of ontlasting.
Het is niet duidelijk waarom bijnierschorsinsufficiëntie en de cerebrale vorm van ALD vrijwel niet voorkomen bij vrouwen met ALD. Het ALD-gen bevindt zich op het X-chromosoom (zie de Feiten over ALD pagina voor meer informatie). Vrouwen hebben twee X-chromosomen. In iedere cel van het lichaam wordt willekeurig één van de twee X-chromosomen geïnactiveerd. Er wordt verondersteld, en laboratorium onderzoek ondersteunt deze hypothese gedeeltelijk, dat bij vrouwen die de cerebrale vorm van ALD ontwikkelen, de X-chromosomen inactivatie niet willekeurig is verlopen. Om de een of andere reden is bij deze vrouwen het X-chromosoom met het gezonde ABCD1-gen juist in alle cellen van het lichaam geïnactiveerd. Dit resulteert in een situatie die vergelijkbaar is met de jongens/mannen met ALD.
Het belang van diagnose
De juiste diagnose van ALD bij vrouwen is van belang voor (1) adequate behandeling, (2) eventuele diagnose en adequate behandeling van kinderen en familieleden en (3) het kunnen aanbieden van onderzoek rondom een eventuele zwangerschap.
De ervaring leert dat de klachten door ruggenmergschade bij vrouwen met ALD vaak lang niet worden herkend. Hierdoor krijgen deze vrouwen niet de specifieke behandelingen die er zijn voor neurologische klachten. Als de diagnose ALD eenmaal gesteld is, kunnen deze klachten makkelijker worden behandeld.
Naast adequate behandeling van de klachten van de vrouw zelf, zorgt een juist diagnose ook ervoor dat eventuele kinderen en familieleden gescreend kunnen worden. Bij mannelijke kinderen en/of familieleden moet gekeken worden naar de aanwezigheid van bijnierschorsinsufficiëntie en cerebrale ALD. Vaak blijken er dan al mannelijke familieleden te zijn met nog niet herkende bijnierschorsinsufficiëntie. Onbehandeld kan bijnierschorsinsufficiëntie resulteren in ernstige complicaties en zelfs de dood. Met hulp van MRI wordt er gekeken naar de aanwezigheid van cerebrale ALD. Indien jongens cerebrale ALD ontwikkelen, kunnen zij behandeld worden met een stamceltransplantatie. Een stamceltransplantatie kan de progressie van cerebrale ALD in jongens en adolescenten stoppen. Een belangrijke voorwaarde is dat de transplantatie in een zeer vroeg stadium van de ziekte wordt uitgevoerd. Het onderzoek naar bijnierschorsinsufficiëntie en cerebrale ALD moet daarom regelmatig herhaald worden.
Tot slot is het van belang om vrouwen te informeren over de mogelijkheden rondom zwangerschap. Tijdens de zwangerschap is het mogelijk om vast te stellen of de foetus een jongen of meisje is door bloedonderzoek bij de ouders. Een vlokkentest of vruchtwaterpunctie kan vaststellen of de foetus ALD heeft. Daarnaast bestaat er de mogelijkheid van preïmplantatie genetische diagnostiek. Hierbij vindt de bevruchting van de eicel buiten de baarmoeder plaats en worden de embryo’s onderzocht op de aanwezigheid van ALD. Alleen embryo’s zonder ALD worden teruggeplaatst.
Dochters van vrouwen met ALD met een kinderwens, kan ook diagnostiek aangeboden worden.
Diagnostiek
De diagnose ALD kan worden gesteld door het aantonen van een verhoogde hoeveelheid verzadigde zeer langketen vetzuren (ZLKV) in het bloed of gekweekte huidcellen. Deze test is een betrouwbare en zeer nauwkeurige methode voor het diagnosticeren van ALD in mannen van alle leeftijden. Maar ongeveer 10-20% van de vrouwen met ALD heeft normale ZLKV in het bloed of in cellen. De test geeft in zo’n geval dus een “fout-negatief” resultaat. Een veelgebruikte manier om “fout-negatieve” resultaten te ondervangen is een combinatie van de ZLKV test met een DNA test. Deze combinatietest maakt het mogelijk om uit te sluiten of een vrouw ALD heeft: normale resultaten voor zowel de biochemische (ZLKV in bloed of huidcellen) als de genetische testen (ABCD1-gen defect bepalen) bevestigen dat een vrouw geen ALD heeft.
Behandeling van klachten
De behandeling van klachten door ruggenmergschade is symptomatisch. Dit betekent dat er geen behandeling is die de zenuwschade kan stoppen of verbeteren. De behandeling is er op gericht de klachten zo dragelijk mogelijk te maken. Eventuele spasticiteit kan worden behandeld met geneesmiddelen die de spierspanning verminderen. Soms zijn injecties met botulinetoxine in aangetaste spieren mogelijk.
Veel vrouwen met ALD ervaren lage rugpijn of pijn in de enkels, knieën en heupen, veroorzaakt door de verhoogde spierspanning in de benen en de daaruit voortvloeiende loopproblemen. Paracetamol en niet-steroïde anti-inflammatoire geneesmiddelen (bijvoorbeeld ibuprofen, naproxen of diclofenac) kunnen effectief zijn bij de behandeling van deze pijn.
Medicatie is beschikbaar om de incontinentie te verminderen, maar als de klachten ernstig zijn werkt dit vaak niet. Incontinentie materialen zijn vaak noodzakelijk.
Een multidisciplinaire aanpak
Vrouwen met ALD met neurologische klachten kunnen baat hebben bij een multidisciplinaire aanpak. De neuroloog kan enkele van de klachten behandelen, maar een verwijzing naar een revalidatiearts of uroloog is vaak noodzakelijk. Indien nodig kan een psycholoog worden geraadpleegd. De klinisch geneticus kan informatie geven over de wijze van overerving van ALD (zie ook Feiten over ALD), de beschikbaarheid van prenatale testen en de noodzaak van familiescreening.
Slotopmerkingen
Veel, zo niet de meeste, vrouwen met ALD zullen neurologische klachten ontwikkelen ten gevolge van schade aan het ruggenmerg en de zenuwen in de benen. Het is belangrijk om deze klachten te herkennen, zodat een effectieve ondersteunende behandeling gestart kan worden. Wanneer de diagnose ALD bij een vrouw wordt gesteld is het belangrijk om de familie te screenen. Vaak leidt dit tot het opsporen van nog niet gediagnosticeerde aangedane familieleden.