

Very long-chain fatty acids
What is the biochemical problem in ALD?
Patients with ALD have an excess of very long chain fatty acids (VLCFA) in most tissues of the body. This excess is most severe in the brain and adrenal glands, causing neurological problems and adrenal dysfunction (Addison’s disease). VLCFAs also accumulate in blood plasma. This makes it possible to diagnose ALD with a blood test.
Normal VLCFA levels in a woman suspected of having ALD. What does this mean?
Plasma VLCFA analysis is the most commonly used diagnostic test for adrenoleukodystrophy. The test relies on the demonstration of elevated levels of C26:0 and increases in the C26:0/C22:0 and C24:0/C22:0 ratios. Experience with this assay in more than 2,000 ALD patients (men and women) has shown that approximately 10 to 15% of women with ALD have normal levels of VLCFA.
Thus, in women, a normal plasma VLCFA level does not exclude the diagnosis of ALD. If a woman is suspected of having ALD, genetic testing is the most reliable method of diagnosis. Ideally, the pathogenic variant in the family must be defined in an affected male or in an obligate heterozygote relative (an obligate heterozygote is either a woman with a father diagnosed with ALD or a woman with two children with genetically proven ALD).
In 2020, it was shown that VLCFA containing C26:0-lysoPC (used in ALD newborn screening) is elevated in all ALD men and >99% of women (to read the open access publication). Even women with ALD with plasma VLCFA levels in the normal range have elevated levels of C26:0-lysoPC in dried blood spots and plasma. Thus, C26:0-lysoPC outperforms VLCFA analysis as a diagnostic biomarker for ALD.
What are fatty Acids?
Before the chemical structure of fatty acids can be explained, a little chemistry background is needed.
Most chemicals we are familiar with, such as salt and water, are made up of different building blocks called “elements”. Salt is made up of the elements sodium (Na) and chlorine (Cl). Its chemical structure is very simple: one atom or molecule of sodium is linked to one atom of chlorine. The force that holds them together is called a chemical bond. Sodium can only bond with one other molecule; the same is true for chlorine. Therefore, the chemical structure of salt can be written as Na-Cl. The line connecting the Na and Cl represents the chemical bond.
Water is a little more complicated. It is made up of the elements hydrogen (H) and oxygen (O). Hydrogen is like Na and Cl in that it can only “bond” with one other element. Oxygen can form two bonds. In water, two H’s bond with one O. Thus, we can write the structure of water as H-O-H. The fact that elements can bond with other elements has allowed nature to produce an incredible, almost limitless number of chemicals that make up us and our environment.
Fatty acids are made up of H, O, and carbon (C). Carbon can form four bonds, making it a very versatile element. The fatty part of fatty acids is a chain of carbon atoms bonded together; each C is also bonded to several H’s.

The “acid” part of a fatty acid has one C, two O’s, and one H.
In the acid part, there are two lines or bonds between the C and one of the O’s. This is called a “double bond”.

Examples of fatty acids
Acetic acid (vinegar), a short-chain fatty acid; C2:0

Palmitic acid (found in lard and butter), a long-chain fatty acid; C16:0

Two very long chain fatty acids (VLCFA’s) are called lignoceric acid; C24:0

and hexacosanoic acid; C26:0

A convenient abbreviation system is also shown with above each fatty acid. The number after the “C” is the number of carbon atoms in the chain, e.g. C2, C16, C24, C26, etc. The number after the colon tells us the number of double bonds in the carbon chain. In all of the examples shown above, there are no double bonds between the carbon atoms, so they all have the designation “:0”.
Fatty acids – What are they for? Where do they come from and what does the body do with them?
Fatty acids are essential chemicals in the body. They are found in more complex chemicals such as triglycerides, phospholipids, sphingolipids, glycolipids, and others. Triglycerides, are the main chemicals in “fat”; they are primarily a storage form. Phospholipids are part of the membranes that surround all cells in the body and the smaller structures inside the cells. Sphingolipids and glycolipids are complex chemicals found primarily in brain and nerve cells; gangliosides and myelin are in this category. The bottom line is that fatty acids are absolutely essential for many normal bodily functions. We couldn’t live without them.
Fatty acids are found in the foods we eat. They are particularly high in fatty or greasy foods, fried foods, and oils. They are also found in large amounts in nuts and seeds. Meat, even lean cuts, is high in fatty acids. On the other hand, vegetables, fruit (except the skin of the fruit), and starchy foods (such as pasta or bread) are relatively low in fatty acids.
What happens to dietary fatty acids? Fatty acids and other nutrients in food enter the stomach, where the process of digestion begins. During this process, food is broken down into its various components, such as carbohydrates (sugars and starches), proteins, and lipids (fatty acids and cholesterol). These food components are then absorbed by the cells that line the intestines. The nutrients pass through the intestinal cells and into small blood vessels. These blood vessels go directly to the liver, which can be thought of as a “processing plant” for nutrients. In the liver cells, nutrients are metabolized, which means they are either broken down to produce heat or energy, or converted into other chemicals that the body needs. Excess nutrients may be converted to a storage form such as glycogen (for sugars) or triglycerides (for fatty acids).
Very often the body has too much fatty acid, either because we have eaten too much or because the body has produced too much internally. We need to get rid of the excess fatty acid. Fatty acids are then broken down or “oxidized” to produce energy or heat for the body. In fact, fatty acids are the main source of fuel for the body during starvation. There is a delicate balance between having enough fatty acids and having too much. The body normally has finely tuned mechanisms to maintain this balance. When the balance is upset, disease often results.
VLCFA’s What do we know about them?
1) Are they normally found in the body?
– YES!
2) What is their function?
– They have many functions. For example, they are integral components of membrane lipids such as sphingolipids and glycerophospholipids. They contribute to the structural integrity and fluidity of cell membranes, which is essential for proper cell function and viability. VLCFAs are essential for the formation and maintenance of the skin barrier. They are important for the synthesis of ceramides, which are essential for the skin’s protective barrier function. VLCFAs are essential for the maintenance of myelin, the protective sheath around nerve fibers. This is critical for proper nerve function and signal transmission.
3) Where do they come from?
– Dietary sources and through production in the body, by elongation of shorter fatty acids into longer fatty acids.
4) What causes the elevated levels of VLCFA in ALD?
– The body makes too much.
– The body doesn’t remove the excess.
5) Which of these possibilities is correct?
– Studies of cells from ALD patients have shown that the process that normally breaks down or oxidizes VLCFAs is defective in ALD.
How does the body normally oxidize fatty acids?
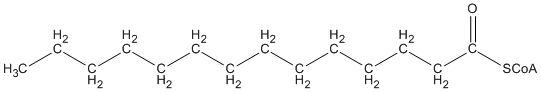
Through a series of chemical reactions, the body shortens fatty acids by removing two carbons at a time:
Fatty acid beta-oxidation

A fatty acid is not very reactive chemically.
The enzyme fatty acyl-CoA synthetase attaches a coenzyme A to the fatty acid.

(“activated fatty acid”)
The enzyme acyl-CoA oxidase adds a “double bond”.

The enzyme enoyl-CoA hydratase uses a water molecule to remove the double bond and insert a hydroxyl (O-H).

The “O-H” is oxidized to “=O” by the enzyme hydroxyacyl-CoA dehydrogenase.

The 16-carbon fatty acid is cleaved by thiolase into a 14-carbon fatty acid and a 2-carbon fatty acid.
 +
+ 
This 14-carbon fatty acyl-CoA can be further shortened by repeating the above process.
The 2-carbon unit can be used for energy production.
How do enzymes work?

Enzymes are proteins in the body that help make chemical reactions happen. Enzymes have “binding sites” for the chemicals with which they interact. This illustration shows the first chemical reaction of fatty acid oxidation. The enzyme has one binding site for a fatty acid and another for a chemical called coenzyme A (CoA). These binding sites are very specific; fatty acid and CoA fit into them like keys into locks. By bringing these two chemicals close together, the enzyme allows them to bond to form a new chemical called fatty acyl-CoA. The enzyme releases this product and is then free to bind another fatty acid and CoA. Without the enzyme, the likelihood of the fatty acid and CoA binding together is almost zero.
What is the “adrenoleukodystrophy protein” and what does it do?

Cells contain small vesicular structures called organelles. This biological compartmentation allows the body to put enzymes that need to work together in the same place. Fatty acid oxidation takes place in two different organelles – the mitochondria and the peroxisomes. The enzymes needed for fatty acid oxidation in the mitochondria are different from those in the peroxisomes. Research conducted at Kennedy Krieger Institute in the 1980s by Drs. Inderjit Singh and Hugo Moser showed that VLCFA oxidation occurs in peroxisomes (Singh et al 1981).
In 1993, Drs. Patrick Aubourg and Jean-Louis Mandel in Paris discovered that the ALD gene produces a protein (the ALD protein or ALDP) that is localized inside cells in the membrane of the peroxisomes (Mosser et al 1993). The ALD protein is a member of a specific family of transporter proteins. These proteins allow molecules to cross biological membranes, such as those surrounding cellular organelles.
ALD is characterized by the inability of cells to metabolize/degrade VLCFA to shorter chain fatty acids. This results in elevated levels of VLCFA in all tissues of the body. The breakdown of VLCFA occurs exclusively in peroxisomes. The enzymes needed to break down VLCFA are functional and present in the peroxisomes of ALD patients. Based on studies showing that restoration of normal ALD protein in patient cells restores VLCFA beta-oxidation (Shinnoh et al 1995) and reduces VLCFA to normal levels (Cartier et al 1995), it has long been hypothesized that the ALD protein transports VLCFA across the peroxisomal membrane. Experiments with yeast cells and cells from ALD patients provided evidence that the ALD protein does indeed transport VLCFA (as VLCFA-CoA) across the peroxisomal membrane (van Roermund et al 2008; Ofman et al 2010).
ALD protein deficiency has two major consequences: 1) it impairs peroxisomal VLCFA beta-oxidation and 2) it increases VLCFA-CoA levels in the cytosol of the cell. These elevated levels of cytosolic VLCFA-CoA provide a substrate for further elongation to even longer fatty acids by ELOVL1, the human C26-specific elongase (Ofman et al 2010; Kemp and Wanders 2010).
The ALD protein
The ALD protein is undetectable by immunofluorescence analysis in approximately 70% of affected individuals. For reasons that are not well understood, the gene product may be absent even in individuals with missense mutations (a single building block of the protein is altered). The main biochemical abnormality is the accumulation of saturated very long-chain fatty acids, especially etracosanoic (C24:0) and hexacosanoic (C26:0) fatty acids, as a result of the impaired ability to degrade these substances, a function that normally takes place in the peroxisome. The ALD protein transports VLCFA from the cytosol to the peroxisome.

Figure: Detection of the ALD protein by immunofluorescence: (left) fibroblasts from a control showing punctate staining indicating the normal presence of the ALD protein in peroxisomes; (middle) fibroblasts from a male patient with ALD and a mutation affecting the stability of the ALD protein. Note that there is no punctate staining; (right) fibroblasts from a female patient with ALD from the same family as the male patient. The ABCD1 gene is located on the X chromosome. Females have two X chromosomes. However, only 1 X chromosome is active in each cell. The cells that show punctate staining are those that have an active copy of the normal ABCD1 gene, while the cells that do not show punctate staining are those that have an active ABCD1 gene that contains the mutation.
Last modified | 2024-06-25