
Thérapie Génique pour l’ALD
Auteurs: Virginie Bonnamain, Ph.D. et Stephan Kemp, Ph.D.
En 2009, les Drs. Nathalie Cartier et Patrick Aubourg et leurs collègues (Hôpital Saint Vincent de Paul, Paris, France) ont publié les résultats positifs d’un traitement par thérapie génique de deux garçons de 7 ans présentant des signes précoces d’une ALD cérébrale (Cartier et al. Science). Ces garçons étaient candidats à une greffe allogénique de cellules souches hématopoïétiques (allo-CSH), mais aucun donneur compatible n’était disponible.
La thérapie génique est basée sur la correction des cellules souches de la moelle osseuse des patients:
- Les cellules souches de la moelle osseuse ont été extraites des patients. Cette procédure s’appelle « aphérèse ».
- En laboratoire, une copie normale du gène ALD a été insérée dans les cellules souches de la moelle osseuse à l’aide d’un virus dérivé du VIH.
- Les patients ont reçu une chimiothérapie pour éradiquer leur moelle osseuse et l’empêcher de produire de nouvelles cellules souches. Cette procédure s’appelle « conditionnement ».
- Ils ont ensuite reçu une infusion intraveineuse de leurs propres cellules souches génétiquement corrigées portant le gène ALD normal.
Film expliquant les bases d’une autogreffe de cellules souches hématopoïétiques (auto-CSH)
https://www.youtube.com/watch?feature=player_embedded&v=NckF2m0SwWo
Film expliquant les bases d’une alloogreffe de cellules souches hématopoïétiques (allo-CSH)
Film réalisé par Sanofi France
Les deux patients ont été suivis pendant 24 et 30 mois, respectivement. Au cours de cette période, 15% de leurs cellules sanguines ont démontré l’expression de la protéine ALD normale, alors qu’avant le traitement, aucune protéine ALD n’était détectable dans leurs cellules sanguines. Le taux d’AGTLC a diminué de 38% dans le plasma des patients.
L’IRM cérébrale et les tests cognitifs ont montré que la progression de la maladie cérébrale s’est arrêtée après 14 à 16 mois. Les patients sont restés stables depuis. La lésion démyélinisante observée dans la voie auditive d’un patient a été réparée. L’arrêt de la démyélinisation cérébrale progressive chez ces deux enfants représente un résultat clinique comparable à celui de l’allo-CSH.
Étude Starbeam
L’étude Starbeam (ALD-102, NCT01896102) est une étude clinique de thérapie génique de phase 2/3 visant à déterminer l’innocuité et la tolérance du traitement de thérapie génique « Lenti-D », et si l’administration d’une dose unique de ce médicament peut arrêter la progression de l’ALD cérébrale. En avril 2018, un total de 31 patients avaient été inclus dans l’étude. Sur ces 31 patients, 29 ont été traités avec Lenti-D et le suivi médian pour tous les patients traités était de 34 mois (0.4 – 54 mois).
Le critère d’évaluation principal de l’efficacité de l’étude Starbeam est l’absence de déficiences fonctionnelles majeures (Major Functional Disabilities – MFD) 24 mois après le traitement. Les MFD correspondent à des incapacités sévères considérées comme ayant un impact fort sur la capacité du patient à fonctionner de manière autonome: perte des capacités de communication, cécité corticale, nécessité de s’alimenter par sonde, incontinence totale, dépendance au fauteuil roulant et perte totale de la mobilité volontaire.
Le critère d’évaluation secondaire de l’efficacité de l’étude est la progression de la maladie cérébrale. Ceci est évalué par la mesure du rehaussement de contraste après injection de gadolinium à l’IRM cérébrale (qui est un indicateur de neuroinflammation) et par le score de la fonction neurologique (NFS). Le NFS est un système de notation utilisé pour mesurer la sévérité des déficits cliniques en évaluant 15 symptômes dans plusieurs domaines.
Le profil d’innocuité de Lenti-D a également été évalué (effets secondaires et analyse génomique de l’intégration du gène).
En octobre 2017, les résultats de 17 patients traités par Lenti-D et ayant terminé les 24 mois de suivi ont été publiés (Eichler et al. 2017). Au moment de l’analyse des données pour ce groupe initial de 17 patients, le suivi médian était de 29.4 mois (21.6 – 42.0 mois).
- Quinze (88%) des 17 patients étaient en vie et présentaient des symptômes cliniques minimes.
- 1 patient est décédé du fait de la progression de la maladie au cours du conditionnement avant le traitement, et
- 1 patient a été retiré de l’étude et est décédé des suites d’une greffe allogénique ultérieure.
- Chez les 15 patients restants, aucune maladie du greffon contre l’hôte n’a été constatée (c’est-à-dire lorsque les cellules greffées « attaquent » les autres cellules du patient).
- Quatorze de ces 15 patients présentaient un score NFS de 0 ou 1, ce qui indique une absence de symptômes cliniques ou des symptômes cliniques minimes.
- Douze des 15 patients avaient un score de Loes stable, indiquant l’absence de progression de la lésion.
- Le rehaussement de contraste au gadolinium, qui était présent au départ chez tous les patients, était absent chez les 15 patients à six mois après la transplantation. Une réémergence du rehaussement de contraste au gadolinium a été constatée chez quelques patients au cours de l’étude (dont 2 patients à 24 mois), mais celui-ci était moins étendu qu’avant le traitement.
Ces premiers résultats suggèrent que la thérapie génique avec Lenti-D pour le traitement de l’ALD cérébrale est au moins aussi efficace qu’une greffe allogénique classique. L’absence de maladie du greffon contre l’hôte indique également que le traitement avec Lenti-D est probablement plus sûr.
Après avoir terminé l’étude ALD-102 (environ 2 ans), les patients ont été inclus dans une nouvelle étude (LTF-304, NCT02698579) pour 13 ans supplémentaires afin d’évaluer l’innocuité et l’efficacité à long terme.
 Figure: Vue d’ensemble du protocole de traitement Starbeam. Image reproduite avec l’aimable autorisation de bluebird bio, et traduite en français
Figure: Vue d’ensemble du protocole de traitement Starbeam. Image reproduite avec l’aimable autorisation de bluebird bio, et traduite en français
Le promoteur de l’étude Starbeam, bluebird bio, est une société de biotechnologies basée à Cambridge, dans le Massachusetts, aux États-Unis
Les faits sur l’ALD
Définition
L’adrénoleucodystrophie (ALD) est une maladie génétique progressive grave qui affecte les glandes surrénales, la moelle épinière et la substance blanche (myéline) du système nerveux. Reconnue en 1923, elle a d’abord été connue sous le nom de maladie de Schilder puis sous celui de leucodystrophie soudanophile. Dans les années 1970, le nom adrénoleucodystrophie a été introduit afin de mieux décrire les manifestations de la maladie. «Adréno» fait référence aux glandes surrénales ; «Leuco» fait référence à la substance blanche du système nerveux et « Dystrophie » signifie croissance anormal ou développement anormal. Il n’y a aucune relation avec l’adrénoleucodystrophie néonatale qui appartient aux troubles de la biogenèse des peroxysomes du spectre de Zellweger.
Biochimie
L’ALD est une maladie héréditaire du stockage métabolique dans lequel un défaut d’une enzyme particulière résulte en l’accumulation d’acides gras à très longue chaîne (AGTLC) dans tous les tissus du corps. Ces AGTLC sont nocifs pour certaines cellules et certains tissus. Pour des raisons qui ne sont pas encore connues, le cerveau, la moelle épinière, les testicules et les glandes surrénales sont principalement touchés. Dans le système nerveux central, l’accumulation des AGTLC finit par détruire la gaine de myéline qui entoure les nerfs, causant des troubles neurologiques. Les AGTLC sont toxiques pour les cellules des glandes surrénales dont le dysfonctionnement provoque la maladie d’Addison (insuffisance surrénale).

Figure 1: les AGTLC qui s’accumulent dans l’ALD sont principalement produits par l’allongement des acides gras à longue chaîne. Pour maintenir l’équilibre fragile de l’homéostasie des AGTLC, les quantités excessives d’AGTLC doivent être dégradées. Les AGTLC ne peuvent être dégradés que dans les peroxysomes. Toutes les cellules du corps, à l’exception des globules rouges, ont des peroxysomes. L’ALD est causée par des mutations dans le gène ABCD1 qui produit la protéine ALDP. L’ALDP transporte les AGTLC du cytosol dans le peroxysome. Une carence en ALDP bloque ce transport, ce qui se traduit par une altération de la dégradation des AGTLC et leur accumulation dans les cellules, tissus et organes. Les enzymes qui sont nécessaires à la dégradation des AGTLC sont présentes dans les peroxysomes, mais les AGTLC ne peuvent pas les atteindre.
Épidémiologie
L’ALD est mondialement présente et ne se limite pas à certains groupes ethniques. L’incidence globale de l’ALD est d’environ 1 nouveau-né sur 15.000.
Génétique
L’ALD est une maladie liée au chromosome X, ce qui signifie que le gène de l’ALD (nom officiel ABCD1) est situé sur le chromosome X. Les hommes ont un chromosome X et un chromosome Y (XY ; Figure 2). Lorsque le père est porteur d’un gène de l’ALD affecté, il n’y a pas d’autre chromosome X pour le protégé ; Par conséquent, il éprouvera des symptômes de l’ALD. Les femmes ont deux chromosomes X (XX; Figure 2). Les femmes chez qui le gène défectueux est présent sont dites « conductrices » car on pensait auparavant que seul un faible pourcentage de femmes conductrices développait des symptômes cliniques. Il est maintenant clair que ce n’est pas le cas (voir ci-dessous et à la page Les femmes ayant une ALD). Les symptômes cliniques chez les femmes sont relativement plus modérés que chez les hommes, cependant, 80% des femmes atteintes d’ALD développent éventuellement des symptômes cliniques. Par conséquent, la terminologie « femmes conductrices » est trompeuse, et ne devrait plus être utilisée. L’explication la plus probable au fait que les femmes développent une forme moins grave de la maladie est la présence d’une copie normale du gène ABCD1 sur l’autre chromosome X. On pense que la présence dans les tissus et les organes, de cellules exprimant la copie saine du gène ABCD1, préserve les femmes ayant une ALD de développer la forme cérébrale (ALD cérébrale).

Figure 2: (A gauche) Si une femme est conductrice du gène défectueux de l’ALD, pour chaque nouveau-né : quand l’enfant est une fille, il y a une chance sur deux que sa fille soit également conductrice de l’ALD et une chance sur deux que sa fille ne soit pas affectée. Si l’enfant est un garçon, il y a 50% de probabilité que le fils soit atteint d’ALD et 50% de chances qu’il ne soit pas affecté. (A droite) Pour les maladies liée à l’X, tels que l’ALD, si un homme touché a des enfants, alors aucun de ses fils n’auront la maladie puisqu’un père transmet toujours son chromosome Y à ses fils. Mais toutes ses filles seront conductrices (il transmet toujours son seul chromosome X (affecté) à ses filles).
L’évolution clinique
Les patients atteints d’ALD sont pré-symptomatiques à la naissance. Chez les hommes, la première manifestation de l’ALD est généralement une insuffisance surrénale, qui peut se produire chez les jeunes bébés. À l’âge adulte, les hommes développent une myélopathie (maladie de la moelle épinière). Les hommes atteints d’ALD peuvent développer une démyélinisation cérébrale progressive (ALD cérébrale), tant dans l’enfance qu’à l’âge adulte. L’ALD cérébral peut être la première manifestation de l’ALD, ou survenir en plus de l’insuffisance surrénale et / ou en plus de la myélopathie (figure 3). Les femmes ayant une ALD sont également affectées et ne sont pas seulement conductrices de la déficience du gène de l’ALD. En effet, à l’âge de 60 ans, plus de 80% de ces femmes développent les signes et symptômes associés à la myélopathie. Les femmes souffrant d’ALD développent rarement une insuffisance surrénale ou une démyélinisation cérébrale.

Figure 3: Le spectre clinique de l’ALD chez l’homme. Les patients ayant une ALD ne présentent aucun symptôme à la naissance. Les barres colorées indiquent l’âge d’apparition possible d’une insuffisance surrénale (barre mauve), d’une myélopathie (mauve pale) et d’une ALD cérébrale (barre grise). L’insuffisance surrénale peut apparaitre dès 5 mois d’âge. À l’âge adulte, les hommes développent systématiquement une myélopathie progressive chronique. Une ALD cérébrale peut débuter à tout âge, le plus jeune des patients décrit ayant 3 ans. Le défaut principal résultant de la mutation du gène de l’ALD et le stockage des AGTLC dans les tissus entraînent une insuffisance surrénale et une myélopathie (appelés ensemble adrénomyéloneuropathie ou AMN). L’apparition d’une ALD cérébrale résulte probablement de la combinaison du défaut principal résultant de la mutation du gène de l’ALD et de différents paramètres environnementaux et / ou génétiques encore inconnus. Il est important de reconnaître que les patients atteints d’insuffisance surrénale et / ou de myélopathie restent exposés au risque de développer une ALD cérébrale.
L’insuffisance surrénale (ou même une crise addisonienne, mettant la vie en danger) peut être le premier symptôme chez les garçons et les hommes atteints d’ALD, des années, voire des décennies, avant l’apparition de symptômes neurologiques. Une étude sur des garçons ALD pré-symptomatiques au niveau neurologique, a montré que 80 % de ces garçons avait déjà une altération de la fonction surrénale au moment du diagnostic de l’ALD. Les signes les plus fréquents d’insuffisance surrénale sont la fatigue chronique ou durable, la faiblesse musculaire, la perte d’appétit, la perte de poids, les douleurs abdominales et les vomissements inexpliqués. D’autres symptômes peuvent inclure des nausées, une diarrhée, une pression artérielle basse (qui diminue encore lorsqu’une personne se lève, provoque des étourdissements ou des évanouissements), l’irritabilité et la dépression, l’envie d’aliments salés, une hypoglycémie, des maux de tête ou des suées. Les individus peuvent ou non avoir une pigmentation cutanée accrue résultant d’une sécrétion excessive d’hormone adrénocorticotrophine (ACTH).
Myélopathie: Pratiquement tous les patients ALD de sexe masculin qui atteignent l’âge adulte développent une myélopathie, typiquement entre 20 et 30 ans. Les symptômes sont limités à la moelle épinière et aux nerfs périphériques. Au début de la maladie le handicap neurologique progresse lentement. Le diagnostic de l’AMN est rarement fait pendant les 3 à 5 premières années des symptômes cliniques, à moins que d’autres cas d’ALD n’aient été identifiés dans la famille. Les patients développent un trouble de la marche progressant lentement, en conséquence de la rigidité et de la faiblesse des jambes. Les individus peuvent également développer un dysfonctionnement de la vessie avec des urgences urinaires, pouvant progresser vers une incontinence complète. Tous les symptômes sont progressifs sur des années ou des décennies, la plupart des patients perdant la marche entre l’âge de 50 et 70 ans.
Adrénomyéloneuropathie (AMN): le terme AMN fait référence aux patients masculins ayant à la fois une fonction surrénale altérée et une myélopathie.
ALD cérébrale: Les garçons et les hommes ayant une ALD présentent le risque de développer des lésions démyélinisantes de la substance blanche du cerveau (ALD cérébrale). L’apparition d’une ALD cérébrale n’a jamais été décrite avant l’âge e trois ans. Jusqu’à présent l’ALD cérébrale était considérée comme rare à l’adolescence (4 à 7%) et chez l’adulte (2 à 5%). Cependant, depuis qu’un large groupe de patients est régulièrement suivit, avec des IRMs réalisées chaque année, il apparait que ces pourcentages sont en fait plus élevés. Aujourd’hui, nous ne pouvons pas prédire si un patient développera ou non une ALD cérébrale, ni quand. Un facteur déclenchant environnemental possible est un traumatisme crânien, mais d’autres facteurs génétiques et environnementaux – aujourd’hui – inconnus sont probablement nécessaires au développement de l’ALD cérébrale. En général, les symptômes de l’ALD cérébrale progressent rapidement. Un nouveau-né garçon a un risque évalué à 35-40 % de développer une ALD cérébrale entre l’âge de 3 ans et 18 ans. Chez les garçons d’âge scolaire, les premiers symptômes sont généralement des problèmes de comportement et des déficits d’apprentissage se manifestant par un déclin de la performance scolaire. Ces premiers symptômes cliniques sont souvent initialement attribués à d’autres troubles tels que le trouble déficitaire de l’attention / hyperactivité, ce qui peut retarder le diagnostic de l’ALD. Chez les patients adultes, les premiers symptômes sont également souvent psychiatriques et peuvent ressembler à une dépression ou une psychose. Chez ces patients, le diagnostic de l’ALD est souvent retardé ; particulièrement s’il n’existe pas d’histoire familiale de la maladie ou si les symptômes surrénaux sont absents. Au fur et à mesure de la progression de la maladie, apparaissent des déficits neurologiques évidents, comme une atteinte auditive et visuelle, la faiblesse des bras et des jambes, des problèmes de coordination et des convulsions. A ce stade, la progression est extrêmement rapide et tout à fait dévastatrice. Les patients affectés peuvent perdre la capacité de comprendre le langage et de se promener en quelques mois. Finalement, les patients sont alités, aveugles, incapables de parler ou de répondre, nécessitant des soins infirmiers à temps plein et une alimentation par tube nasogastrique ou par gastrostomie. Le décès se produit généralement 2 à 4 ans après le début des symptômes initiaux de l’ALD cérébrale, ou – si les soins sont importants – les patients peuvent rester dans cet état végétatif apparent pendant des années.
Les femmes ayant une ALD: Comme dans de nombreuses maladies liées à l’X, il était supposé initialement que la majorité des femmes atteintes d’ALD ne présentaient pas de symptômes. Cependant, il est aujourd’hui établi que cette notion est fausse. En réalité, plus de 80% des femmes atteintes d’ALD de 60 ans ont développé des symptômes (voir également à la page Les femmes ayant une ALD). Le texte intégral de l’étude décrivant les symptômes chez les femmes ayant une ALD peut être consulté et téléchargé (en format pdf). En général, l’apparition des premiers symptômes neurologiques se produit à un âge plus avancé que chez les hommes atteints d’une myélopathie ; généralement entre 40 et 50 ans. Le handicap moteur et la progression de la maladie sont généralement moins graves que chez l’homme. De façon singulière, et contrairement aux hommes, l’incontinence fécale est une plainte fréquente chez les femmes atteintes d’ALD. Il est important de noter que la myélopathie chez les femmes atteintes d’ALD est souvent diagnostiquée à tort comme une sclérose en plaques. L’insuffisance surrénale et l’ALD cérébrale sont très rares, présentes chacune dans moins de 1% des cas (voir la page Les femmes ayant une ALD pour plus de détails).
Test
L’ALD est diagnostiquée par un simple test sanguin, au cours duquel est évaluée la quantité d’acides gras à très longue chaîne. Ce test est fiable chez les hommes et est largement reconnu comme un moyen très précis de diagnostiquer les hommes de tous âges. Cependant, pour environ 20% des femmes ayant une ALD, le test des AGTLC montre des résultats normaux correspondant à des «faux négatifs». Une façon d’identifier avec précision les patients «faux négatifs» est par un test d’ADN. Ce test de laboratoire permet une identification précise des femmes ayant une ALD en testant leurs gènes, et quand ces résultats sont normaux, peuvent garantir à une femme qu’elle n’est pas conductrice d’un gène ALD muté.
Le dépistage néonatal
Le diagnostic précoce de l’ALD est la clé pour sauver des vies, car le dépistage néonatal permet une surveillance prospective de la fonction surrénale et de l’apparition de l’ALD cérébrale. Un test de dépistage des nouveau-nés a été développé. Il permet de détecter une élévation des AGTLC (C26:0 – LysoPC) dans des échantillons sanguins. Le 30 décembre 2013, l’état de New York a lancé le dépistage néonatal de l’ALD. En février 2016, l’ALD a été ajouté au Panel de dépistage uniforme recommandé des États-Unis (RUSP). Depuis, d’autres États et pays ont lancé des programmes de dépistage néonatal ou ont entrepris des processus visant à ajouter l’ALD à leurs programmes de dépistage néonatal existants. Des informations détaillées et à jour sur le dépistage néonatal de l’ALD peuvent être trouvées à la page “dépistage néonatal“.
Recherche
Des recherches approfondies sur l’ALD sont conduites dans le monde entier. En 1993, le gène de l’ALD a été identifié grâce aux efforts combinés des Drs. Patrick Aubourg et Jean-Louis Mandel en France et du Dr. Hugo Moser aux États-Unis. Cette découverte a ouvert des portes à de nouvelles études plus approfondies. Les recherches sont axées sur de nombreux aspects et tentent de répondre à des questions fondamentales, comme: « Comment les AGTLC peuvent éventuellement entraîner la perte de myéline ? » ; « Pourquoi un patient développe une ALD cérébrale, tandis qu’un autre (qui peut même être son frère) développe une myélopathie à un âge plus avancé ? ».
Traitement
Il n’existe pas aujourd’hui de traitement curatif de l’ALD.
Traitement hormonal de substitution des stéroïdes surrénaliens: la plupart des patients ALD masculins développent une insuffisance surrénale. L’insuffisance surrénale est souvent la première manifestation de l’ALD: une étude approfondie a révélé que 80% des garçons ayant une ALD sans symptômes neurologiques, identifiés lors d’un dépistage familiale élargi, avaient déjà une fonction surrénalienne altérée depuis l’âge de 4 ans. Pour ces patients, une traitement hormonal de substitution des stéroïdes surrénaliens est obligatoire et peut leur sauver la vie, bien qu’une gestion réussie du dysfonctionnement surrénalien n’ait aucun effet sur les symptômes neurologiques.
Pour la myélopathie, qui affecte 85 % de tous les patients ALD (hommes et femmes combinés), aucun traitement curatif n’est disponible.
Restriction alimentaire: Parce que les AGTLC sont toxiques pour la myéline, les glandes surrénales et les testicules, plusieurs tentatives ont été menées afin de diminuer les concentrations plasmatiques d’AGTLC. La restriction alimentaire d’AGTLC seule n’a pas d’effet sur les concentrations plasmatiques d’AGTLC.
L’huile de Lorenzo: Les AGTLC sont principalement synthétisés par l’allongement de chaîne d’acides gras plus courts. En laboratoire, l’addition d’acides gras mono-insaturés dans le milieu de culture de fibroblastes ALD réduit les concentrations d’AGTLC à des niveaux normaux. Ceci peut être expliqué car les enzymes nécessaires à la synthèse de AGTLC sont les mêmes pour les acides gras mono-insaturés et pour les acides gras saturés. Mais leur affinité pour les acides gras mono-insaturés est plus élevée. Cette constatation a servi de base à une approche diététique. L’administration orale d’acide oléique sous forme de triglycérides (GTO) et l’acide érucique sous forme de triglycérides (GTE) normalise les taux plasmatiques d’AGTLC en 1 mois chez la plupart des patients atteints d’ALD. La combinaison de GTO et de GTE dans un rapport de 4: 1 est connue sous le nom de « l’huile de Lorenzo », en hommage à Lorenzo Odone, le premier patient traité avec ce mélange. On pensait que l’huile de Lorenzo était très prometteuse. Cependant, plusieurs études ouvertes ont montré que l’huile de Lorenzo n’améliore pas la fonction neurologique ou endocrinienne ni qu’elle pouvait arrêter la progression de la maladie (voir la page sur l’huile de Lorenzo pour plus de détails).
Lovastatine: Un effet de la lovastatine a été montré sur les AGTLC. Cet effet n’a cependant pas pu être reproduit par d’autres équipes. Des expériences ultérieures ont en fait montré que les statines n’ont aucun effet sur les niveaux d’AGTLC dans le cerveau ni dans les surrénales chez la souris ALD, et auraient même provoqué l’accumulation d’AGTLC dans ces tissus. En raison de ces résultats contradictoires, un essai clinique en double aveugle randomisé et contrôlé par placebo pour tester l’effet de la lovastatine comme une thérapie diminuant les AGTLC dans l’ALD a été effectuée au Centre médical universitaire d’Amsterdam. Les résultats et les conclusions démontrent que le traitement par la lovastatine entraine une petite diminution des AGTLC dans le plasma, mais n’a aucun effet sur les niveaux d’AGTLC dans les cellules, puisque les niveaux de C26:0 étaient stables dans les globules rouges et blancs.
Bézafibrate: Dans la recherche de composés qui pourraient réduire les niveaux d’AGTLC, le bézafibrate, un médicament utilisé pour le traitement de l’hyperlipidémie, a été identifié comme un agent diminuant les AGTLC. Des expériences ont montré que dans les fibroblastes, le bézafibrate réduit les AGTLC en inhibant directement l’activité de l’élongase ELOVL1, spécifique des AGTLC. Une étude ouverte pilote a été réalisée pour évaluer l’effet du bézafibrate sur l’accumulation d’AGTLC dans les cellules sanguines des patients AMN. Malheureusement, le bézafibrate ne diminua pas les AGTLC dans les cellules sanguines de patients ALD. Cela était très probablement dû à son incapacité à atteindre des concentrations thérapeutiques adéquates chez les patients.
Greffe de moelle osseuse: Chez les garçons et les adolescents à un stade précoce d’ALD cérébrale, la greffe allogénique de cellules souches hématopoïétiques (GCSH) peut arrêter la progression de la démyélinisation cérébrale due à l’ALD, à condition que la greffe soit effectuée à un stade très précoce de la maladie. L’efficacité de la GCSH est basée sur le renouvellement des cellules microgliales cérébrales déficientes en ALDP par des cellules microgliales normales qui proviennent des cellules souches de la moelle osseuse du donneur. (Voir la page Hematopoietic stem cell transplantation (HSCT) pour plus de détails).
La thérapie génique: Il est envisagé que dans un avenir relativement proche, la transplantation de cellules souches hématopoïétiques autologues (provenant du patient lui-même) génétiquement corrigées ex vivo (à l’extérieur du corps du patient) à l’aide d’un vecteur lentiviral, puis re-perfusées, pourrait devenir une option thérapeutique supplémentaire, sur la base des résultats très encourageants rapportés en 2009 chez les deux premiers patients ALD traités, et suite aux récents résultats de l’étude Starbeam publiés en octobre 2017 (Voir la page Thérapie Génique pour l’ALD pour plus de détails).
A 10 minute overview of ALD
Produced by Youreka Science in collaboration with ALD Connect, Inc.
Please see the ALD Connect Educational Videos & Webinars page for more videos
Acides Gras à Très Longue Chaîne
Au plan biochimique, quel est le problème dans l’ALD / AMN ?
Chez les patients ayant une ALD ou une AMN, il y a trop d’acides gras à très longues chaines (AGTLC) qui s’accumulent dans la plupart des tissus du corps. Cet excès est particulièrement grave dans le cerveau et les glandes surrénales et il en résulte des problèmes neurologiques et un dysfonctionnement de la glande surrénale (maladie d’Addison). Les AGTLC s’accumulent également dans le plasma sanguin, ce qui permet un diagnostic de l’ALD par un test sanguin.
Des niveaux normaux d’AGTLC chez une femme soupçonnée d’avoir une ALD. Qu’est-ce que ça veut dire ?
L’analyse des taux d’AGTLC plasmatiques est le test diagnostic le plus fréquemment utilisé pour diagnostiquer une ALD. Le test est basé sur la démonstration d’une augmentation des niveaux de C26:0 et de l’augmentation des rapports C26:0/C22:0 et C24:0/C22:0. L’expérience a montré que chez plus de 2000 patients ALD testés (hommes et femmes), près de 10 à 20% des femmes atteintes d’ALD avaient des niveaux normaux d’AGTLC.
Un niveau d’AGTLC plasmatique normal n’exclut donc pas l’ALD du diagnostic. Quand une femme est soupçonnée d’être porteuse d’une ALD, l’analyse de la mutation est la méthode la plus fiable pour le diagnostic. Idéalement, la mutation dans la famille doit être identifiée chez un parent affecté ou une parente porteuse hétérozygote avérée (une parente porteuse hétérozygote avérée est, soit une femme dont le père a été diagnostiqué avec une ALD, soit une femme ayant eu deux enfants atteints d’une ALD génétiquement prouvée).
Que sont les acides gras?
Pour comprendre la structure chimique des acides gras, quelques notions de chimie sont nécessaires.
La plupart des produits chimiques que nous connaissons, comme le sel et l’eau, sont constitués de différentes briques appelées des «éléments». Le sel est constitué d’éléments sodium (Na) et chlore (Cl). Sa structure chimique est très simple: un atome ou une molécule de sodium est lié à un atome de chlore. La force qui les maintient ensemble est appelée une liaison chimique. Le sodium a la capacité de se lier avec une autre molécule; la même chose est vraie pour le chlore. Par conséquent, la structure chimique du sel peut être écrite sous forme de Na-Cl. Le trait reliant le Na et le Cl représente la liaison chimique.
L’eau est un peu plus compliquée. Elle est composée des éléments hydrogène (H) et oxygène (O). L’hydrogène, comme le Na et le Cl, ne peut se «lier» qu’avec un autre élément. L’oxygène, lui, peut former deux liaisons. Dans l’eau, deux H se lient avec un O. Ainsi, on peut écrire la structure de l’eau de la façon suivante H-O-H. Le fait que les éléments peuvent se lier avec d’autres éléments a permis à la nature de produire un nombre incroyable, presque illimité, de produits chimiques qui nous composent, nous ainsi que notre environnement.
Les acides gras sont composés de H, O, et de carbone (C). Chaque carbone peut former quatre liaisons; ce qui fait du carbone un élément très polyvalent. La partie grasse des acides gras est une chaîne d’atomes de carbone liés ensemble; chaque C étant également lié à plusieurs H.

La partie “acide ” d’un acide gras a un C, deux O et un H.
Dans la partie acide, il y a deux traits ou liaisons entre l’extrémité C et l’un des O. c’est ce qu’on appelle une « double liaison ».

Des exemples d’acides gras
L’acide acétique (vinaigre), est un acide gras à chaîne courte; C2:0

L’acide palmitique (trouvé dans le saindoux et le beurre), est un acide gras à chaîne longue; C16:0

Deux acides gras à très longue chaîne (AGTLC): l’acide lignocérique; C24:0

et l’acide hexacosanoïque; C26:0

Un système pratique d’abréviation est associé à chaque acide gras ci-dessus. Le nombre après le «C» représente le nombre d’atomes de carbone dans la chaîne, par exemple C2, C16, C24, C26, etc. Le nombre après les deux points nous indique le nombre de doubles liaisons dans la chaîne carbonée. Dans tous les exemples indiqués ci-dessus, il n’y a pas de double liaison entre les atomes de carbone; Par conséquent, tous ont la désignation « :0 ».
Les acides gras – Qu’ont-ils de bon ? D’où viennent- ils et qu’en fait le corps ?
Les acides gras sont des produits chimiques nécessaires au corps. Ils se retrouvent dans des produits chimiques plus complexes, tels que des triglycérides, des phospholipides, des sphingolipides, des glycolipides et d’autres. Les triglycérides sont les principaux produits chimiques du “gras”; ils permettent avant tout le stockage. Les phospholipides font partie des membranes qui entourent toutes les cellules du corps et les petites structures à l’intérieur des cellules. Les sphingolipides et les glycolipides sont des produits chimiques complexes que l’on trouve principalement dans les cellules du cerveau et du système nerveux ; les gangliosides et la myéline font partie de cette catégorie. Le point essentiel est que les acides gras sont absolument nécessaires pour de nombreuses fonctions normales du corps. Nous ne pouvons pas vivre sans eux.
Les acides gras se trouvent dans les aliments que nous mangeons. Ils sont en quantités particulièrement élevées dans les aliments gras, les graisses, les aliments frits et les huiles. Ils sont également présents en grandes quantités dans les noix et les graines. La viande, même dans les viandes maigres, contient beaucoup d’acides gras. D’un autre coté les légumes, les fruits (sauf la peau des fruits), et les féculents (par exemple les pâtes ou le pain) sont relativement faibles en acides gras.
Que deviennent les acides gras alimentaires ? Les acides gras et les autres nutriments présents dans les aliments atteignent l’estomac où la digestion commence. Les aliments sont alors scindés en leurs différents composants, tels que les hydrates de carbone (sucres et amidons), les protéines et les lipides (des acides gras et cholestérol). Ces composants de l’alimentation sont ensuite absorbés par les cellules qui tapissent l’intestin. Les nutriments passent par les cellules intestinales et entrent dans les petits vaisseaux sanguins. Ces vaisseaux sanguins conduisent directement au foie, qui peut être considéré comme une «usine de traitement» des éléments nutritifs. Dans les cellules hépatiques, les éléments nutritifs sont métabolisés ; ce qui signifie qu’ils sont, soit décomposés pour produire de la chaleur ou de l’énergie, soit convertis en d’autres produits chimiques dont le corps a besoin. Des nutriments en excès peuvent être transformés sous une forme permettant le stockage comme le glycogène (pour les sucres) ou les triglycérides (pour les acides gras).
Très souvent le corps a trop d’acide gras, soit parce que nous avons trop mangé, soit parce que le corps en fabrique trop en interne. Nous avons besoin de nous débarrasser de l’excès d’acide gras. Les acides gras sont alors décomposés ou “oxydés” pour produire de l’énergie ou de la chaleur pour le corps. En effet, les acides gras sont la principale source de carburant pour le corps en cas de famine. Il y a un équilibre fragile entre avoir assez d’acide gras et en avoir trop. Normalement le corps a des mécanismes sensibles qui maintiennent cet équilibre. Lorsque l’équilibre est rompu, survient souvent une maladie.
Les AGTLC : Que savons-nous à leur sujet ?
1) Sont-ils normalement présents dans le corps ?
– OUI!
2) Quelle est leur fonction?
– Ils font partie des membranes présentent dans le cerveau, y compris de la myéline, « l’isolant » autour des fibres nerveuses.
3) D’où viennent-ils?
– Ils proviennent de l’alimentation et de leur production dans le corps, par l’élongation des acides gras plus courts en acides gras plus long
4) Que pourrait causer l’augmentation des niveaux d’AGTLC dans l’ALD/AMN ?
– Le corps en fabrique trop
– Le corps n’élimine pas les excédents
5) Laquelle de ces possibilités est correcte?
– Des études menées chez des patients volontaires et dans des cellules en laboratoire ont montré que le processus qui élimine ou oxyde habituellement les AGTLC est défectueux dans l’ALD/AMN.
Comment le corps oxyde-t-il normalement les acides gras ?
Grâce à une série de réactions chimiques, le corps raccourcie les acides gras en enlevant deux par deux les atomes de carbone:
Béta-oxydation des acide gras

Un acide gras est chimiquement peu réactif.
L’enzyme acyl-CoA synthétase associe une Coenzyme A à un acide gras.

(« Acide gras activé »)
L’enzyme acyl-CoA oxydase insère une « double liaison ».

L’enzyme énoyl-CoA hydratase utilise une molécule d’eau pour éliminer la double liaison et d’insérer un hydroxyle (O-H).

« O-H » est oxydé en « =O » par l’enzyme hydroxyacyl–CoA déshydrogénase.

L’acide gras à 16 carbones est coupé par l’acétyl-CoA acétyltransférase (thiolase) pour donner un acide gras à 14 carbones et un acide gras à 2-carbones.
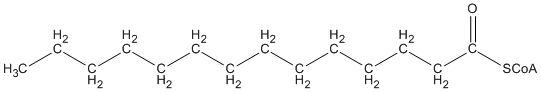 +
+ 
Cet acyl-CoA à 14 carbones peut encore être réduit en répétant le processus ci-dessus.
L’unité de 2 carbones peut être utilisée pour la production d’énergie.
Comment fonctionnent les enzymes ?

Les enzymes sont des protéines dans le corps qui facilitent les réactions chimiques. Les enzymes ont des « sites de liaison » pour les produits chimiques avec lesquels elles interagissent. Dans le schéma ci-dessus, la première réaction chimique de l’oxydation des acides gras est représentée. L’enzyme a un site de liaison pour un acide gras et un autre pour un produit chimique appelé le coenzyme A (CoA). Ces sites de liaison sont très spécifiques ; l’acide gras et le CoA se logent dans leur site de liaison respectif comme des clés dans des serrures. En rapprochant ces deux composés chimiques l’un de l’autre, l’enzyme leur permet de se lier pour former un nouveau produit chimique appelé un acyl–CoA. L’enzyme libère ce produit, et redevient libre de se lier à d’autres acides gras et CoA. Sans l’enzyme, la probabilité d’associer un acide gras à un CoA est quasiment nulle.
Qu’est-ce que la «protéine ALD » et que fait-elle ?

Les cellules contiennent des petites structures vésiculaires appelées organites. Le compartimentage biologique permet à l’organisme de regrouper dans un même lieu les enzymes qui ont besoin de travailler ensemble. L’oxydation des acides gras a lieu dans deux organites différents – les mitochondries et les peroxysomes. Les enzymes nécessaires à l’oxydation des acides gras dans les mitochondries sont différentes de celles utilisées dans les peroxysomes. Les recherches menées à l’Institut Kennedy Krieger dans les années 1980 par les Drs. Inderjit Singh et Hugo Moser ont montré que l’oxydation des AGTLC a lieu uniquement dans les peroxysomes (Singh et al, 1981).
En 1993 à Paris, les Drs. Patrick Aubourg et Jean-Louis Mandel ont découvert que le gène ALD génère une protéine (ALDP) qui se situe à l’intérieur des cellules dans la membrane des peroxysomes (Mosser et al, 1993). La protéine ALD appartient à une famille spécifique de protéines de transport. Ces protéines permettent à des molécules de traverser les membranes biologiques comme celles entourant les organites cellulaires.
L’ALD est caractérisée par l’incapacité des cellules à métaboliser/dégrader les AGTLC en acides gras à chaîne plus courte. Il en résulte des taux d’AGTLC élevés dans tous les tissus du corps. La dégradation des AGTLC a lieu exclusivement dans les peroxysomes. Les enzymes qui sont nécessaires à la dégradation des AGTLC sont fonctionnelles et présentes à l’intérieur des peroxysomes chez les patients ALD. Sur la base des études qui démontrent qu’une expression normale de la protéine ALD dans les cellules de patients atteint d’ALD restaure la bêta-oxydation des AGTLC (Shinnoh et al 1995) et réduit les taux d’AGTLC à des niveaux normaux (Cartier et al, 1995), il a longtemps été émis l’hypothèse que l’ALDP transportait les AGTLC à travers la membrane des peroxysomes. Des expériences utilisant des cellules de levure et des cellules provenant de patients ALD ont fourni les preuves que l’ALDP transporte effectivement les AGTLC (comme AGTLC -CoA) à travers la membrane des peroxysomes (van Roermund et al, 2008 ; Ofman et al, 2010).
Le déficit de l’ALDP dans l’ALD a deux conséquences majeures : 1) il empêche la bêta-oxydation des AGTLC dans les peroxysomes et 2) il induit une augmentation des taux d’AGTLC-CoA dans le cytosol de la cellule. Ces niveaux élevés d’AGTLC-CoA dans le cytosol sont sujets à un allongement supplémentaire par ELOVL1, l’élongase humaine spécifique des acides gras en C26 (Ofman et al, 2010; Kemp et Wanders, 2010).
ALDP
La protéine ALD (ALDP) n’est pas détectable par immunofluorescence chez environ 70 % des personnes touchées. Pour des raisons qui ne sont pas bien comprises, le produit du gène peut être absent même chez les personnes qui ont des mutations faux-sens. L’anomalie biochimique principale est l’accumulation d’acides gras saturés à très longue chaîne, en particulier l’acide hexacosanoïque (C26:0) et l’acide tétracosanoïque (C24:0), dont la cause est l’impossibilité de les dégrader, cette fonction se déroulant normalement dans le peroxysome. La protéine ALDP transporte les AGTLC du cytosol vers le peroxysome.

Figure: Détection de l’ALDP par immunofluorescence. (À gauche) fibroblastes contrôles montrant une coloration en points reflétant la présence normale d’ALDP dans les peroxysomes. (Centre) fibroblastes provenant d’un patient ayant une ALD avec une mutation affectant la stabilité de l’ALDP. Notez qu’il n’y a pas de coloration en points mais une coloration diffuse. (À droite) fibroblastes provenant d’une patiente ayant une ALD de la même famille que le patient masculin au centre. Le gène ABCD1 est situé sur le chromosome X. Les femelles ont deux chromosomes X. Cependant, dans chaque cellule, seulement 1 chromosome X est actif. Les cellules qui présentent une coloration en points sont celles qui ont une copie active du gène ABCD1 normal, tandis que celles qui ne présentent pas de coloration en points sont les cellules qui ont un gène actif ABCD1 porteur de la mutation.
Les images ont été faites par le Dr. Merel Ebberink
Dépistage néonatal
Introduction
A la naissance, les nouveau-nés ALD sont normaux sur le plan neurologique. Cependant, le diagnostic précoce chez les garçons atteints d’ALD peut permettre une intervention parfois vitale. Cette intervention peut prendre la forme d’une mise en place au moment opportun d’un traitement de substitution enzymatique des stéroïdes surrénaliens quand l’insuffisance surrénalienne est détectée, et d’une greffe allogénique de cellules souches hématopoïétiques (allo-CSH) comme traitement de l’ALD cérébrale. Une greffe allo-CSH peut arrêter la progression, souvent fatale, de la démyélinisation cérébrale, à condition que la procédure soit réalisée à un stade très précoce de la maladie. Malheureusement, cela n’est efficace que dans une fenêtre thérapeutique étroite, souvent déjà passée. Le dépistage néonatal permet d’agir dans cette « fenêtre d’opportunité » et permet l’initiation rapide de ces thérapies reconnues.
En février 2016, l’ALD a été ajoutée au Panel de dépistages recommandés au niveau fédéral (RUSP) aux États-Unis, qui liste toutes les maladies génétiques pour lesquelles un programme de dépistage chez les nouveau-nés est recommandé au niveau fédéral. L’état de New York a commencé le dépistage de l’ALD chez les nouveau-nés le 30 décembre 2013. Depuis, d’autres états américains ont commencé le dépistage néonatal de l’ALD (Fig 1). Aux États-Unis, plusieurs autres États ont reçu l’approbation législative. En dehors des États-Unis, le ministre de la Santé aux Pays-Bas a approuvé l’ajout de l’ALD au programme de dépistage chez les nouveau-nés. L’initiation du dépistage néonatal de l’ALD est attendue dans ces états américains et ce pays, dès que les ressources budgétaires, les procédures de test et les protocoles de suivi seront en place.

Figure 1: Carte montrant les états qui ont initié le dépistage néonatal de l‘ALD.
Critères d’inclusion d’une maladie dans le programme de dépistage
Il existe un large consensus international sur les critères d’inclusion d’une maladie dans un programme de dépistage chez les nouveau-nés.
- Le diagnostic précoce doit apporter un avantage direct pour le nouveau-né. Il doit y avoir des gains substantiels sur l’état de santé, résultants d’une intervention précoce, dans des maladies graves dont l’histoire naturelle est connue.
- Le test de dépistage doit être de bonne qualité. Le test de mesure doit avoir une spécificité et une sensibilité élevées, c’est-à-dire des taux très faible de faux positifs et de faux négatifs.
 Histoire
Histoire
En 2004, lors de la réunion du Comité consultatif national américain pour le dépistage néonatal, le Dr Hugo Moser a proposé l’ajout de l’ALD au RUSP des États-Unis. Le seul problème était qu’un test valide pour le dépistage néonatal n’était pas disponible. Pour y remédier, il a recueilli des fonds et recruté une équipe de chercheurs au Kennedy Krieger Institute (Baltimore, Maryland) pour identifier un biomarqueur approprié et développer un test utilisant la spectrométrie de masse en tandem (MS / MS). En 2006, l’équipe a détecté la présence C26:0-lysophosphatidylcholine (C26: 0-LPC) dans les taches de sang séché collectées chez les nouveau-nés garçons atteint d’ALD (Hubbard et al. 2006). Dans les années qui ont suivi, les scientifiques ont continué à perfectionner l’analyse (Hubbard et al. 2009; Theda et al. 2014). En collaboration avec des chercheurs de la Mayo Clinic (Rochester, Minnesota), une méthode à haut débit pour l’analyse de C26:0-LPC a ensuite été développée (Haynes and De Jesús 2012; Turgeon et al. 2015). En 2013, cette méthode a été validée à partir de 100 000 taches de sang séché anonymes.

La loi d’Aidan (Aidan’s Law)
En avril 2012, à la suite du décès de leur fils, Aidan, atteint d’ALD cérébrale, qui avait été diagnostiqué trop tard, la famille Seeger rédigea et appuya l’adoption de la loi dite d’Aidan dans l’état de New York. Le projet de loi a été approuvé en février 2013 et est devenu loi en mars 2013. Le 30 décembre 2013, le laboratoire de dépistage néonatal de l’État de New York a commencé à tester les nouveau-nés pour l’ALD.
État de New York
Au cours des trois premières années, l’État de New York a dépisté plus de 700 000 nouveau-nés et identifié 45 enfants avec une ALD: 22 garçons et 23 filles. Sur la base de ces chiffres, l’incidence de l’ALD à la naissance est de 1 sur 15 000. Lorsqu’un nouveau-né atteint d’ALD est identifié, le médecin de famille est avisé et un généticien clinique est sollicité pour confirmer le diagnostic, et permettre un conseil génétique pour le soutien et le dépistage de l’ALD chez les autres membres de la famille à risque (dépistage de la famille étendue).
Pour les garçons, il est impératif d’initier une surveillance séquentielle par IRM cérébrale pour détecter les toutes premières traces du développement d’une ALD cérébrale, et amorcer des tests de la fonction surrénale pour détecter l’insuffisance surrénalienne. Une évaluation complète des fonctions neurologiques, neuropsychologiques, neuroradiologiques et surrénaliennes est nécessaire car il n’existe pas de test pour prédire le développement clinique particulier d’un enfant né avec une mutation ALD.
Le test de dépistage du nouveau-né
Le test C26:0-LPC et les réactifs utilisés peuvent différer légèrement d’un laboratoire à l’autre. Cependant, dans tous les cas, le diagnostic de l’ALD est réalisé en utilisant un processus à trois niveaux (Fig 2). Le premier niveau est une analyse MS / MS standard à haut débit de C26:0-LPC. Les échantillons qui ont une concentration en C26:0-LPC élevée sont ensuite testés à un deuxième niveau, en utilisant un test HPLC-MS / MS. Ce test est plus spécifique, mais prend aussi plus de temps. Pour les échantillons qui présentent toujours un C26:0-LPC élevé, le séquençage du gène ABCD1 est réalisé dans un troisième temps.

Figure 2: Le principe du dépistage de l’ALD à 3 niveaux.
Défis
Des défis importants existent dans plusieurs pays et des discussions éthiques sont en cours concernant la mise en place du dépistage néonatal de l’ALD.
- Le premier critère permettant l’ajout d’une maladie à un programme de dépistage néonatal, qui requière que le diagnostic précoce apporte un avantage direct pour le nouveau-né, peut être à l’origine de préoccupations éthiques. Dans l’ALD, environ un tiers des garçons développeront une ALD cérébrale entre l’âge de 3 et 18 ans. Cependant, les deux tiers restants des hommes ALD développeront une adrénomyéloneuropathie (AMN) à l’âge adulte, caractérisée par une spasticité des membres, des troubles de la marche et une incontinence. L’AMN est traitée de façon symptomatique. L’absence de marqueurs de laboratoire ou d’autres outils biologiques, fait qu’il est difficile de prédire les effets de la maladie sur la santé des individus, ce qui peut augmenter le risque d’interventions médicales inutiles.
- Le dépistage néonatal identifie également les filles portant un gène ALD défectueux. Les femmes atteintes d’ALD ont moins de 1% de risque de développer une insuffisance surrénalienne ou une ALD cérébrale, et il n’y a donc aucun avantage direct pour la santé d’une nouveau-née avec ALD puisqu’elle ne sera pas traitée par greffe allo-CSH ou hormonothérapie surrénalienne. Environ 80% des femmes atteintes d’ALD développeront une myélopathie à l’âge de 60 ans.
- Dans plusieurs pays, un débat grandit au sein de la communauté scientifique et parmi les organisations de patients, sur l’inclusion de certaines maladies intraitables dans les programmes de dépistage. Comme mentionné précédemment, le diagnostic précoce doit finalement aboutir à un bénéfice direct pour la santé du nouveau-né lui-même. Cela peut ne pas être évident dans le cas d’une maladie qui est diagnostiquée, mais n’est pas encore traitable.
- Parfois, le dépistage néonatal identifie des maladies dépassant le cadre du test prévu. Le test de dépistage néonatal de C26:0-LPC identifie également des maladies intraitables associées à une augmentation des niveaux de C26:0-LPC (Fig 2). Notamment, les troubles du spectre de Zellweger, des troubles de l’oxydation des acides gras péroxysomaux causés par un défaut de l’acyl-CoA oxydase 1 (ACOX1) péroxysomale ou d’une protéine multifonctionnelle (HSD17B4), acyl-CoA binding domain containing protein 5 (ACBD5) deficiency, le syndrome de délétion contiguë ABCD1 DXS1357E (CADDS), et le syndrome d’Aicardi-Goutières.
- Dans certains cas, d’autres avantages, allant au-delà de ceux qui visent manifestement à améliorer la santé du nouveau-né, peuvent être envisagés. Certains d’entre eux peuvent bénéficier au nouveau-né lui-même, comme un parcours de diagnostic plus rapide. Mais la plupart des avantages concernent la famille. Notamment la possibilité d’un dépistage familial élargi qui permet d’identifier d’autres membres de la famille à risque et de s’adapter aux conséquences de la maladie sur la vie de la famille. Enfin, les parents peuvent également bénéficier du dépistage d’une maladie pour laquelle il n’existe pas de traitement efficace, car cette information peut être prise en compte dans leurs projets d’enfants. Cependant, il existe également des inconvénients évidents. Le diagnostic d’une maladie incurable peut jeter une ombre ou stigmatiser l’enfance et la vie du nouveau-né.
En raison de son ajout au RUSP américain, le lancement du dépistage néonatal de l’ALD est attendu dans les années à venir dans un nombre croissant d’États aux États-Unis, et d’autres pays devraient suivre. Ces mesures permettront clairement d’améliorer le parcours clinique de centaines de nouveau-nés ALD, de leurs parents et de leurs proches.
Présentations cliniques
L’adrénoleucodystrophie liée à l’X (ALD) est une maladie qui peut se présenter sous de nombreuses formes différentes (phénotypes). Les symptômes vont d’une maladie progressive de la moelle épinière chez les hommes et les femmes (AMN) à une maladie du cerveau dont l’issue est fatale chez les garçons et les hommes (ALD cérébrale). L’adrénoleucodystrophie est causée par un défaut génétique (mutation) du gène ABCD1. Une mutation du gène ABCD1 est présente chez tous les patients ALD. Plus de 700 mutations différentes ont été identifiées jusqu’à présent – la plupart d’entre elles étant répertoriées dans cette base de données. Malheureusement, les mutations n’ont aucune valeur prédictive quant à la gravité de la maladie chez un individu particulier. Bien que tous les bébés nés avec une ALD aient une mutation dans le gène ABCD1, l’évolution clinique d’un patient individuel reste entièrement imprévisible, même parmi les membres d’une famille qui sont porteurs de la même mutation. Jusqu’à présent, aucun marqueur prédictif n’a pu être identifié; ni les taux d’acides gras à très longue chaîne (AGTLC), ni la mutation du gène, ni l’histoire familiale relative à l’adrénoleucodystrophie, ni la présence ou l’absence de la protéine ALD dans des lignées cellulaires en culture.
Prévalence de l’ALD
L’incidence globale de l’ALD est d’environ 1 nouveau-né sur 14.000. L’ALD a été identifiée dans tous les pays européens et d’Amérique latine, en Chine, au Japon, en Inde, en Israël, en Arabie Saoudite, ainsi que dans tous les groupes ethniques, y compris les Afro- Américains, les Amérindiens et les Maoris.
Les femmes ayant une ALD
Pré-symptomatique: L’ALD est démontrée par l’un des critères suivants : niveaux élevés d’AGTLC, mutation identifiée dans le gène ABCD1, père ayant une ALD, ou naissance d’au moins deux enfants atteints d’ALD. Le nombre de femmes ayant une ALD pré-symptomatiques diminue avec l’âge. La majorité des femmes de moins de 30 ans n’ont pas de symptôme neurologique.
Nous recommandons d’informer les femmes à risque d’être hétérozygotes pour l’ALD et en âge de procréation, sur la disponibilité des tests génétiques. Elles peuvent alors décider de façon éclairée si elles souhaitent faire un test et également se voir proposer de prendre contact avec un conseiller en génétique.
Myélopathie: Comme dans de nombreuses maladies liées à l’X, il a été supposé initialement que la majorité des femmes atteintes d’ALD ne présentaient pas de symptôme. Cependant, dans une étude récente, il a été montré que plus de 80% des femmes atteintes d’ALD développent des symptômes après l’âge de 60 ans. Le texte intégral de cette étude peut être consulté et téléchargé (en format pdf). En général, l’apparition des premiers symptômes neurologiques se produit à un âge plus avancé que chez les hommes atteints d’ALD; généralement entre 40 et 50 ans (figure 1). Le handicap moteur et la progression de la maladie sont généralement moins graves, mais certaines femmes atteintes d’ALD sont aussi sévèrement touchées que les patients de sexe masculin. Il est important de noter que les femmes atteintes d’ALD sont souvent diagnostiquées à tort comme ayant une sclérose en plaques. Bien qu’il y ait beaucoup de femmes touchées par l’ALD, beaucoup d’entre elles ne sont pas diagnostiquées car elles restent sans symptômes pour la plupart jusqu’à un âge moyen. De plus, de nombreuses femmes présentant des symptômes demeurent non diagnostiquées à moins qu’elles n’aient un parent de sexe masculin symptomatique. Une étude de l’Institut Kennedy Krieger a démontré que sur 616 familles identifiées avec une ALD, dans seulement 16 cas le premier patient identifié dans la famille était une femme. La grande majorité des patientes ALD ne sont seulement identifiées qu’après qu’un homme de leur famille ait été diagnostiqué (par le biais des programmes de dépistage familiaux). La meilleure méthode pour établir qu’une femme a une ALD est l’analyse génétique du gène ABCD1. Mais pour plus de fiabilité, la mutation dans la famille doit être identifiée chez un parent masculin malade ou une parente porteuse de la mutation. Une femme porteuse est une femme qui soit a un père diagnostiqué avec une ALD, soit a deux enfants atteints d’ALD dont le diagnostic a été génétiquement prouvé.
Nous recommandons que les femmes à risque de porter une mutation responsable d’une ALD, quand elles sont en âge de procréer, soient informées de la disponibilité des tests génétiques. Elles peuvent alors décider de façon éclairée si elles souhaitent faire un test et également se voir proposer de prendre contact avec un conseiller en génétique.
Implication cérébrale: une implication cérébrale est très rarement observée chez les femmes ayant une ALD, seuls quelques cas isolés sont connus.
Insuffisance surrénale primaire cliniquement évidente: une insuffisance surrénale, appelée aussi maladie d’Addison (voir ci-dessous), est très rare chez les femmes ayant une ALD quel que soit leur âge, cela représente moins de 1% des patientes.

Figure 1: Corrélation entre l’état symptomatique et l’âge, dans une cohorte de 46 femmes atteintes d’ALD. En vert, le pourcentage de femmes au sein de chaque tranche d’âge ayant développé des symptômes cliniques liés à l’ALD. Les points noirs indiquent chaque individu de la cohorte, classé comme étant soit symptomatique soit pré-symptomatique.
Les hommes ayant une ALD
Pré-symptomatique: les patients pré-symptomatiques, en particulier ceux identifiés lors d’un dépistage dans leur famille, ne présentent pas d’anomalies neurologiques ou endocriniennes. L’ALD est diagnostiquée par l’un des critères suivants: mise en évidence de taux élevés d’AGTLC ou d’une mutation identifiée dans le gène ABCD1. Mais chez ces patients il n’y a aucun indice d’une atteinte surrénale ou neurologique. Le nombre d’hommes pré-symptomatiques diminue avec l’âge. Il est important que le médecin évalue ces patients fréquemment. Le suivi des garçons et des hommes avec une ALD pré-symptomatiques est important pour deux raisons: 1) il permet la détection précoce d’une insuffisance surrénale, et 2) il permet la détection précoce d’une ALD cérébrale par IRM et de proposer une greffe de cellules souches hématopoïétiques (GCSH) allogénique si un donneur compatible ou du sang de cordon sont disponibles. Le risque est élevé pour ces patients de développer des symptômes neurologiques ou des anomalies endocriniennes. Une ALD pré-symptomatique est fréquente chez les garçons de moins de 3 ans et très rare chez les hommes de plus de 40 ans.
Insuffisance surrénale: Une évaluation prospective de la fonction surrénalienne chez 49 garçons ayant une ALD neurologiquement pré-symptomatique a montré que 80 % d’entre eux présentaient déjà une altération de la fonction surrénale. Une insuffisance surrénale non diagnostiquée est connue pour être une cause fréquente de morbidité et peut être évitée par une surveillance attentive, l’identification précoce de l’altération de la réserve surrénalienne, et la mise en place en temps utile d’un traitement hormonal de substitution. C’est souvent la première manifestation d’une ALD, souvent avant l’apparition des symptômes neurologiques. Les signes les plus courants d’une insuffisance surrénale sont une fatigue chronique ou de longue durée, une faiblesse musculaire, une perte d’appétit, une perte de poids, des douleurs abdominales et des vomissements inexpliqués. D’autres symptômes peuvent inclure des nausées, des diarrhées, une pression artérielle basse (qui tombe encore plus bas quand une personne se lève, causant des étourdissements ou des évanouissements), l’irritabilité et la dépression, une envie d’aliments salés, une hypoglycémie, ou une diminution de sucre dans le sang, des maux de tête, ou de la transpiration. Les individus peuvent ou non avoir une augmentation de la pigmentation de la peau due à la sécrétion excessive de l’hormone adrénocorticotrophine (ACTH).
ALD cérébrale (infantile, de l’adolescent et de l’adulte): Les symptômes d’une ALD cérébrale sont en général rapidement progressifs. Le type de symptômes développés dépend de l’âge d’apparition de la maladie. L’ALD cérébrale se produit souvent dans la petite enfance (ALD cérébrale infantile; CCALD), mais jamais avant l’âge de 3 ans. Généralement les garçons touchés ont d’abord des problèmes de comportement ou des troubles d’apprentissage, souvent diagnostiqués comme un trouble du déficit de l’attention, avec ou sans hyperactivité. Ces comportements peuvent persister pendant des mois ou plus, mais sont suivis par des symptômes évocateurs d’un trouble sous-jacent plus grave. Ces symptômes peuvent inclure un décrochement à l’école : inattention, détérioration des aptitudes à l’écriture, baisse des résultats scolaires; difficulté de compréhension orale (bien que la perception sonore soit normale); difficulté à la lecture, d’orientation spatiale, de compréhension des documents écrits ; maladresses; troubles visuels et parfois une vision double ; et un comportement agressif ou désinhibé. Un examen d’IRM cérébrale réalisé à ce moment peut être étonnamment anormale même lorsque les symptômes sont relativement bénins. Chez certains garçons, des convulsions peuvent en être la première manifestation. Le taux de progression est variable. Il peut être extrêmement rapide avec une incapacité totale après six mois à deux ans, qui conduit au décès à des échéances variables. La plupart des individus ont déjà une altération de la fonction surrénale au moment où les premiers troubles neurologiques sont détectés. Un patient nouveau-né a un risque de 35-40 % de développer une ALD cérébrale entre l’âge de 3 ans et 18 ans, mais la forme cérébrale peut aussi se déclencher à l’âge adulte. Chez les patients adultes, la présentation et la progression des ALD cérébrales sont identiques à celles observées chez l’enfant, avec des changements de comportement et / ou des symptômes psychiatriques. Les adultes qui développent une ALD cérébrale ont déjà souvent des signes d’une AMN et d’une insuffisance surrénale.
Adrenomyeloneuropathy (AMN): Pratiquement tous les patients atteints d’ALD qui atteignent l’âge adulte développent une AMN. Les symptômes sont limités à la moelle épinière et aux nerfs périphériques. Le diagnostic d’une AMN est rarement fait pendant les 3-5 premières années des symptômes cliniques, à moins que d’autres cas d’ALD aient été identifiés dans la même famille. La présentation typique est un homme d’une vingtaine d’années ou d’âge moyen qui développe une raideur progressive et une faiblesse des jambes, un déficit de la sensibilité aux vibrations dans les membres inférieurs, des troubles sphinctériens, et une impuissance. Tous ces symptômes progressent sur des décennies. Les cheveux des patients atteints d’AMN sont souvent fins et clairsemés; ces patients présentent souvent une calvitie précoce vers l’âge de vingt ans. La plupart des hommes ayant une AMN ont déjà une altération de la fonction surrénale au moment où les premiers troubles neurologiques sont détectés. Les hommes ayant une AMN peuvent également développer une ALD cérébrale. Le risque exact n’est pas connu et l’apparition d’une ALD cérébrale est imprévisible. Le risque de développer une ALD cérébrale secondaire à une AMN est estimé à au moins 20% sur une période de 10 ans.
Les femmes ayant une ALD
Pendant de nombreuses années l’ALD a été considérée comme une maladie ne touchant que les garçons et les hommes. Il est aujourd’hui évident que les femmes porteuses développent également des symptômes neurologiques. Exceptionnellement, les femmes ayant une ALD présentent des déficiences surrénaliennes ou une forme cérébrale de la maladie. Les femmes ayant une ALD sont, par conséquent, un groupe distinct de patients ALD. Cette page traite des complaintes et des symptômes que peuvent développer les femmes ayant une ALD, et de ce qui peut y être fait.
Introduction
Contrairement aux concepts initiaux, il a été clairement démontré que les troubles liés au chromosome X peuvent affecter non seulement les hommes, mais aussi dans une moindre mesure, les femmes porteuses des gènes défectueux. L’ALD n’est pas une exception. Les femmes ayant une ALD sont aussi des patientes, mais cela est moins reconnu. Souvent, les symptômes sont attribués à d’autres causes, et donc un traitement symptomatique peut être retardé. Dans notre expérience, aux Pays-Bas, les femmes avec une ALD non diagnostiquée avaient subi des interventions chirurgicales (remplacement de la hanche, opérations de la colonne cervicale), parfois à plusieurs reprises, pour ce qui s’est révélé être plus tard des symptômes liés à la moelle épinière, conséquents à une ALD. Pire encore, ne pas diagnostiquer une femme porteuse d’une ALD empêche de réaliser un conseil génétique et de poser un diagnostic et un traitement approprié aux autres membres de la famille.
Histoire d’une femme porteuse
Quand elle avait 38 ans, Mme H., née aux Pays-Bas, vivait aux Etats-Unis avec sa famille. Elle avait remarqué qu’elle avait de plus en plus de mal à maintenir son équilibre. En outre, elle a commencé à sentir ses jambes un peu raides. Elle a eu des problèmes de fonction de la vessie pendant plusieurs années, mais supposait que d’avoir donné naissance à ses deux enfants en était la cause. Un de ses cousins, chez qui ont avait d’abord suspecté une tumeur au cerveau, est mort à l’âge de 13 ans. Un autre cousin, qui avait des problèmes progressifs de marche, a été diagnostiqué avec une sclérose en plaques. Après avoir lu un article sur l’ALD dans un magazine populaire en 1985, elle a reconnu ses symptômes et ceux de ses cousins, et a décidé de contacter le Dr Hugo Moser au Kennedy Krieger Institute de Baltimore, MD. Un échantillon de sang a été prélevé et l’analyse a révélé une élévation des acides gras à très longue chaîne (AGTLC), confirmant le diagnostic d’une ALD.
Lorsque Mme H. a eu 55 ans, elle a visité le Centre médical universitaire d’Amsterdam, aux Pays-Bas. Il est apparu que certains de ses proches avaient été répertoriés dans la littérature médicale, mais apparemment beaucoup n’avaient pas encore été évalués concernant l’ALD. Certains ont choisi de ne pas l’être, mais beaucoup ne s’étaient jamais vu proposer une analyse des AGTLC. À l’examen physique, le tonus musculaire dans les jambes était augmenté, et il y avait une certaine faiblesse musculaire. Les sensations étaient altérées : le tact fin, les vibrations et la sensation de posture étaient tous altérés dans les jambes et les pieds. Les réflexes tendineux étaient brusques dans les jambes, et les réflexes plantaires étaient en extensions.
Les imageries par résonance magnétique (IRM) du cerveau et de la moelle épinière étaient normales. Un conseil génétique et des tests sanguins ont été proposés aux parents.
Rétrospectivement, des ALD cérébrales infantiles et des AMN ont été diagnostiqués chez ses cousins. Elle a été orientée vers un médecin spécialiste en réadaptation et vers un physiothérapeute.
Avec son mari, directeur de l’organisation néerlandaise de patients, elle a soutenu depuis de nombreuses années les familles et les patients récemment diagnostiqués. Jusqu’à ce qu’elle décède subitement et de manière inattendue, d’une maladie sans rapport avec l’ALD.
Les symptômes chez les femmes porteuses d’une ALD
Depuis les années 1980, il est établit que les femmes ayant une ALD peuvent développer des symptômes. Seulement récemment, une grande étude systématique a été réalisée pour étudier ce phénomène. Dans une étude menée à l’AMC aux Pays-Bas, il a été démontré qu’avec l’âge, la fréquence des femmes ayant une ALD symptomatique augmente (Figure 1). Plus de 80% des femmes atteintes d’ALD auront développé des symptômes à 60 ans.
Figure 1: Corrélation entre l’état symptomatique et l’âge, dans une cohorte de 46 femmes atteintes d’ALD. En vert, le pourcentage de femmes au sein de chaque tranche d’âge ayant développé des symptômes cliniques liés à l’ALD. Les points noirs indiquent chaque individu de la cohorte, classé comme étant soit symptomatique soit pré-symptomatique.
Il est très important de souligner que les symptômes sont extrêmement rares dans l’enfance. De plus, chez les femmes âgées ayant une atteinte de la moelle épinière, les formes cérébrales sont extrêmement rares. Les symptômes chez les femmes touchées sont principalement des anomalies de la moelle épinière et des nerfs des jambes, identiques à ceux de l’AMN. Au fil des décennies se développent une faiblesse et une spasticité des jambes, des sensations perturbées des membres inférieurs, et un contrôle réduit de la vessie et de l’intestin. Contrairement aux hommes affectés, il est très rare que les femmes développent une insuffisance surrénale, bien que dans un large groupe cela ait été évalué à 1 % des cas. Aucune des femmes porteuses néerlandaises qui ont été étudiées ne présentait des signes de dysfonctionnement des glandes surrénales
On ne sait pas pourquoi les femmes ayant une ALD ne développent pas la forme cérébrale. Le gène de l’ALD est situé sur le chromosome X (pour plus d’informations voir à la page : Les faits sur l’ALD). Normalement, l’un des deux chromosomes X présents dans les cellules de chaque femme est inactivé au hasard. L’hypothèse émise et partiellement soutenue par des études en laboratoire, est que chez les femmes qui développent la forme cérébrale de l’ALD les chromosomes X ne sont pas inactivées de manière aléatoire, mais que pour une raison quelconque le chromosome X normal, celui qui n’a pas la mutation du gène ABCD1, est inactivé dans toutes les cellules. Il en résulte une situation similaire à celle des garçons ayant une ALD cérébrale.
Pourquoi les femmes porteuses doivent être identifiées
Beaucoup de femmes porteuses présentant des symptômes bénins d’AMN ne sont pas reconnus pendant de nombreuses années. Cela peut entraîner une absence de traitement spécifique de la spasticité ou des douleurs aux articulations et dans le bas du dos, des dysfonctionnements de la vessie ou de l’intestin. Une fois le diagnostic posé, ces symptômes peuvent être traités plus facilement. Plus important encore, une fois qu’une femme est diagnostiquée comme porteuse d’ALD, ses enfants et ses parents peuvent faire un test afin de diagnostiquer les garçons et les hommes ayant une insuffisance surrénalienne. En outre, le dépistage prénatal (à l’aide du liquide amniotique ou d’une biopsie de trophoblaste) permet l’identification précoce d’un fœtus atteint. Les garçons et les hommes qui sont touchés et qui ont une dysfonction encore méconnue des glandes surrénales peuvent être identifié. Non traitée, l’insuffisance surrénalienne peut entraîner des complications graves et même le décès. Les garçons touchés par la maladie peuvent être surveillés de près ; quand ils développent des symptômes cérébraux, ils peuvent subir une greffe de moelle osseuse ou de cellules souches. Le bénéfice est prouvé chez les garçons ayant une forme cérébrale légère. Les filles de femmes porteuses qui désirent avoir des enfants peuvent également se voir proposer des tests d’AGTLC et d’ADN.
Établir le diagnostic
L’ALD était classiquement diagnostiquée par l’analyse des AGTLC dans les échantillons de sang ou de cellules de peau en culture (fibroblastes). Chez les garçons et les hommes, un niveau anormalement élevé d’AGTLC dans le plasma ou dans les fibroblastes en culture confirme le diagnostic d’une ALD. Mais chez au moins 10 à 15% des femmes atteintes d’une ALD, les AGTLC sont dans les limites normales. Apparemment, leurs chromosomes X non touchés ont suffisamment d’activité résiduelle et sont capables de masquer le déficit biochimique. Par conséquent, dans une famille touchée par la maladie, il est impossible d’exclure qu’une femme n’ait pas une ALD, sur la base des concentrations d’AGTLC normales dans le sang ou dans les cellules en culture! A l’inverse, quand les niveaux d’AGTLC sont élevés, cela confirme le diagnostic.
De nos jours, le diagnostic par l’ADN (analyse de mutation) est la norme pour l’identification des femmes atteintes d’ALD ou soupçonnées d’avoir une ALD. Dans les familles touchées par la maladie, le test ADN ne devrait jamais être omis chez les femmes pour lesquelles les taux d’AGTLC sont dans les limites normales. Jusqu’à présent, plus de 700 mutations différentes dans le gène ABCD1 ont été identifiées. L’analyse de l’ADN peut être réalisée à partir du sang ou d’autres échantillons de tissus, y compris les biopsies et les cellules des villosités choriales, du liquide amniotique ou de cellules déjà en culture.
Stratégie diagnostique
Lorsque qu’une ALD est diagnostiquée dans une famille, il faut souligner combien il est important que tous les membres de la famille fassent un test. Cela peut empêcher l’apparition de nouveaux cas inutiles, et aider à identifier les parents ayant une insuffisance surrénale et ceux qui peuvent bénéficier d’une greffe de moelle osseuse ou de cellules souches. Pour que l’identification des parents touchés soit plus facile et plus fiable, l’analyse des AGTLC ainsi que l’analyse de l’ADN doivent être effectuées dans une famille nouvellement diagnostiquée avec une ALD. Une fois que la mutation a été trouvée, l’analyse mutationnelle dans la famille est beaucoup plus facile, car le test peut être focalisé sur cette anomalie spécifique. L’analyse de l’ADN doit être effectuée dans des centres spécialisés.
Dérèglement hormonal
Bien que le dérèglement endocrinien ait été décrit chez les femmes ayant une ALD, il reste très rare. Environ 1% de ces femmes présentent des signes d’altération de la fonction des glandes surrénales. Une fonction anormale de la glande thyroïde a été rapportée, mais est rencontrée fréquemment dans la population globale, ce qui rend difficile de conclure à une relation avec l’ALD. En général, les tests endocriniens ne devraient être effectués que lorsqu’une insuffisance endocrinienne est suspectée.
Neuro-imagerie et électrophysiologie
Très occasionnellement, de graves anomalies de la substance blanche chez les femmes ayant une ALD ont été décrites. Une implication cérébrale suspectée peut être visualisée par IRM. Des examens électrophysiologiques, comme l’électroencéphalogramme (EEG) ou un test de réponse à un potentiel évoqué « exogène », peuvent aussi être anormaux. Quand les nerfs périphériques sont impliqués, les études de conduction nerveuse et d’électromyographies (EMG) peuvent aussi être anormales.
A des fins de recherche, ces enquêtes sont intéressantes, mais il est essentiel de ne pas procéder à ces tests chez chaque femme porteuse, à moins qu’il n’existe des anomalies clairement identifiées lors de l’examen physique qui justifient une enquête plus approfondie.
Traitement et gestion de la maladie
Traitement curatif
Au cours des dernières décennies, de nombreux traitements ont été testés dans l’ALD. L’huile de Lorenzo, les interférons ß, des traitements par immunoglobulines en intraveineuses, l’immunosuppression avec des médicaments cytostatiques, et l’échange thérapeutique de plasma n’ont jusqu’à présent pas été en mesure d’arrêter ou de retarder la progression de la maladie lorsque les symptômes neurologiques étaient présents. Pour le moment, la greffe de moelle osseuse ou de cellules souches chez les garçons ayant une atteinte cérébrale légère est le seul traitement qui ait montré un bénéfice.
Cependant, chez les hommes et les femmes avec des symptômes de type AMN, une greffe de moelle osseuse ou de cellules souches ne doit pas être effectuée, car ce traitement agressif agit sur l’inflammation cérébrale, et non pas sur la perte lente et progressive des fonctions des cellules nerveuses, dans la moelle épinière et les nerfs périphériques, observée dans l’AMN et chez les femmes porteuses.
Traitement symptomatique
La spasticité peut être traitée avec des médicaments qui réduisent le tonus musculaire ou parfois des injections avec de la toxine botulique dans les muscles touchés.
Beaucoup de femmes ayant une X-ALD font l’expérience de douleurs lombaires ou dans les chevilles, les genoux et les hanches, causées par le tonus musculaire accrue dans les jambes et la marche anormale qui en découle. Les analgésiques non opioïdes, tels que l’acétaminophène et les médicaments anti-inflammatoires non stéroïdiens (par exemple l’ibuprofène, le naproxène ou le diclofénac) peuvent être particulièrement utiles dans le traitement de la douleur.
Des médicaments sont disponibles pour réduire les incontinences urinaires impérieuses, mais souvent cela n’est pas efficace si les symptômes sont graves. Du matériel de protection de l’incontinence est souvent nécessaires.
Une approche multidisciplinaire
Les femmes avec une ALD ayant des symptômes neurologiques peuvent bénéficier d’une approche multidisciplinaire. Le neurologue peut traiter certains des symptômes, mais le renvoi à un médecin spécialiste en médecine physique et de réadaptation, ou à un urologue, est souvent nécessaire. Si nécessaire, un psychologue peut également être consulté pour aider les patientes à faire face.
Enfin et surtout, le généticien clinique ne doit pas être oublié, dans la mesure où le mode de transmission des ALD, la disponibilité des tests prénataux, et la nécessité du dépistage des membres de la famille doivent être discutés.
Remarques pour conclure
Beaucoup, sinon la plupart des femmes atteintes d’ALD, développeront des symptômes neurologiques causés par des dommages de la moelle épinière et des nerfs périphériques. Il est important de reconnaître ces symptômes pour offrir un traitement symptomatique efficace. Il faut garder à l’esprit qu’à l’heure actuelle des outils très puissants et fiables de diagnostic sont disponibles pour démontrer ou exclure qu’une femme est porteuse d’une ALD. Il est important de souligner que la norme pour le diagnostic chez les femmes est l’analyse de l’ADN, et que le test des AGTLC peut très bien être normal chez 10 à 15% des femmes porteuses. Une fois que le diagnostic de l’ALD a été établi, la famille doit faire des tests et les parents masculins touchés doivent être identifiés. Ces tests et le dépistage de la famille doivent être effectués de préférence dans des centres spécialisés.
Histoire de l’ALD
1910: Rétrospectivement, Haberfeld et Spieler firent dès 1910 la première description clinique d’un patient atteint d’une adrénoleucodystrophie liée à l’X (Haberfeld et Spieler, 1910). Un garçon de 6 ans en bonne santé développa une peau profondément bronzé (hyperpigmentation), une acuité visuelle diminuée, et une baisse de son niveau scolaire. Dans les mois qui suivirent, ce garçon devint incontinent, perdit le langage et développa une tétraparésie spastique, qui progressa finalement vers une perte de la marche. Il fut hospitalisé à l’âge de 7 ans, et décéda 8 mois plus tard. Son frère aîné était mort d’une maladie semblable à l’âge de 8 ans. Un examen histologique du cerveau post mortem révéla d’importants changements dans la substance blanche du cerveau, combinée à une accumulation péri-vasculaire de lymphocytes et des cellules plasmatiques dans le système nerveux, indiquant une réponse inflammatoire.
1923: Siemerling et Creutzfeld ont rapporté le cas d’un garçon avec une progression similaire de la maladie, comprenant une peau bronzée et des résultats d’examens neuropathologiques comme décrits par Haberfeld & Spieler en 1910, mais avec également une atrophie du cortex surrénalien.
1963: A cette époque, neuf cas comparables avaient été signalés. Le fait que tous les patients soient des hommes suggéra une transmission récessive liée à l’X (Fanconi et al, 1963).
1970: Le nom adrénoleucodystrophie vient de l’association caractéristique d’une leucodystrophie avec une insuffisance surrénalienne primaire (adrénocorticale) (Blaw, 1970).
1972: Le point de départ de toute les découvertes ultérieures sur la maladie a été l’observation faite par Powers, Schaumburg, et Johnson que les cellules surrénales des patients atteint d’ALD contenaient des inclusions lipidiques caractéristiques (gouttelettes de graisse), suivi par la démonstration que ces gouttelettes de graisse consistaient en esters de cholestérol contenant un excès surprenant et caractéristique d’acides gras à très longue chaîne (AGTLC).
1976: Une forme de la maladie progressant plus lentement et caractérisée par une insuffisance surrénalienne, une myélopathie et une neuropathie périphérique, a été décrite chez l’adulte (Budka et al, 1976). Un an plus tard, cinq autres cas ont été signalés par Griffin et al. qui a proposé que cette présentation clinique de l’ALD soit nommée adrénomyéloneuropathie (AMN) (Griffin et al. 1977; Schaumburg et al.1977).
1981: L’identification des AGTLC comme biomarqueurs de l’ALD a conduit à la mise au point d’un test de diagnostic pour l’ALD basé sur la mise en évidence des taux élevés d’ AGTLC dans les cellules de la peau en culture (fibroblastes), dans le plasma, dans les globules rouges et dans les amniocytes (Moser et al. 1981). Ces tests permettent un diagnostic postnatal et prénatal précis. Des études métaboliques ont démontré que les AGTLC sont métabolisés (par bêta-oxydation) exclusivement dans des organites intracellulaires appelés peroxysomes et que cette oxydation des AGTLC est diminuée dans les fibroblastes des patients atteints d’ALD (Singh et al 1981). Par conséquent, l’ALD est une maladie péroxysomale.
1981: Le locus de l’ALD a été localisé sur l’extrémité du bras long du chromosome X, en Xq28 (Migeon et al. 1981).
1982: La première greffe de moelle osseuse (GMO) a été réalisée chez un garçon ayant une ALD cérébrale. Une GMO allogénique prélevée chez son frère, donneur sain HLA identique, a été réalisée chez un garçon de 13 ans ayant une ALD progressant rapidement. La greffe et la récupération hématologique complète ont eu lieu en 4 semaines. Dix jours après la GMO, les taux d’AGTLC et l’activité enzymatique des globules blancs sont devenus normaux ; après 3 mois, il y a eu une réduction progressive des taux plasmatiques d’AGTLC légèrement supérieurs à la normale. Mais la détérioration neurologique a continué. Le patient est décédé d’une infection à adénovirus 141 jours après la GMO.
1986: Rizzo et al. ont démontré que l’addition d’acide oléique (C18:1) dans le milieu de culture normalise les niveaux d’AGTLC saturés dans des fibroblastes cutanés provenant de patients ALD mis en culture. Ces résultats ont servi de base pour le développement de l’huile de Lorenzo. Le traitement des patients atteints d’ALD avec l’huile de Lorenzo normalise les taux plasmatiques en AGTLC en 4 semaines (Moser et al. 1987). Plusieurs essais ouverts ont montré que l’huile de Lorenzo ne permet pas d’améliorer les fonctions neurologiques ou endocriniennes ni d’arrêter la progression de la maladie. Malheureusement, l’efficacité clinique de l’huile de Lorenzo n’a jamais été évaluée dans un essai clinique contrôlé de façon appropriée par placebo. En 2001, le professeur Hugo Moser a écrit: «Notre point de vue est que la thérapie par l’huile de Lorenzo ne se justifie pas chez la plupart des patients qui ont déjà des symptômes neurologiques. Le bénéfice clinique de l’huile de Lorenzo est au mieux limité”.
1990: L’équipe du Pr. Patrick Aubourg a réalisé la première greffe de moelle osseuse (GMO) réussie (Aubourg et al, 1990). Ils ont greffé un garçon de 8 ans présentant des signes neurologiques et neuropsychologiques modérés, et des anomalies modérées en IRM. Son faux jumeau sain était le donneur. Le patient a complètement récupéré et les anomalies neurologiques, neuropsychologiques, et mises en évidence à l’IRM, ont disparues. Lorsqu’elle est effectuée au stade le plus précoce de la démyélinisation cérébrale, une greffe de cellules de moelle osseuse ou de cellules souches hématopoïétiques (GCSH) peut stabiliser ou même inverser la démyélinisation cérébrale chez les garçons ou les adolescents atteints d’ALD.
1993: Une équipe dirigée par les Drs. Mandel et Aubourg a identifié par clonage positionnel le gène responsable de l’ALD (ABCD1) (Mosser et al. 1993). L’identification du gène de l’ALD a permis la détection des mutations responsables de la maladie, le diagnostic prénatal et l’identification des porteurs sains de la maladie.
1997: Trois laboratoires ont décrit la génération d’un modèle murin d’ALD (Forss-Petter et al. 1997; Kobayashi et al. 1997; Lu et al. 1997). Alors que la souris ALD présente les mêmes anomalies biochimiques que celles observées chez les patients, elle ne développe pas une ALD (Pujol et al. 2002).
1999: La base de données ALD a été créée par Hugo Moser et Stephan Kemp. Initialement, cette base a uniquement servi comme registre de toutes les mutations identifiées dans le gène ABCD1, mais elle a été rapidement élargie pour fournir des informations sur de nombreux aspects de l’ALD.
2001: Il a été rapporté et établi que l’ALD affecte tous les groupes ethniques et qu’il s’agit du trouble péroxysomal le plus courant avec une incidence estimée à 1: 17.000 (hommes et femmes confondus) (Bezman et al. 2001). Cela fait de l’ALD la leucodystrophie héréditaire la plus fréquente.
2005: Biochimiquement, l’ALD est non seulement caractérisée par un défaut de la dégradation des AGTLC dans les peroxysomes, mais aussi par une augmentation de l’allongement ultérieur de la chaîne des AGTLC (Kemp et al. 2005).
2006: L’équipe dirigée par le Dr Ann Moser a développé une méthode d’analyse à haut débit des AGTLC (utilisant C26:0 – LysoPC comme métabolite diagnostic) utilisable sur les taches de sang séché (Hubbard et al. 2006). Ces progrès dans le dépistage des AGTLC permettent d’ajouter l’ALD aux programmes de dépistage néonataux.
2009: Une équipe dirigée par les Drs. Cartier et Aubourg a traité avec succès par thérapie génique deux garçons de 7 ans ayant des signes précoces d’ALD cérébrale (Cartier et al. 2009). Les scans cérébraux en IRM et les tests cognitifs ont montré un arrêt de la progression de la maladie cérébrale 14 à 16 mois après le traitement. Ce qui est comparable aux résultats cliniques de la GCSH.
2010: L’équipe de recherche du Dr. Stephan Kemp a établi que l’ALDP transporte les AGTLC à travers la membrane des peroxysomes. Une carence en ALDP a deux effets majeurs : d’une part elle porte atteinte à la dégradation des AGTLC dans les peroxysomes, et d’autre part elle augmente les niveaux cytosoliques d’AGTLC. Ces AGTLC sont alors encore plus allongés par ELOVL1, l’élongase humaine spécifique des C26 (Ofman et al. 2010).
2014: Aux États-Unis, l’état de New York initie le dépistage néonatal de l’ALD (Vogel et al. 2015). Le diagnostic précoce de l’ALD est la clé pour sauver des vies, car le dépistage néonatal permet une surveillance prospective et une intervention précoce.
2015: Aux États-Unis, le Connecticut introduit le dépistage néonatal de l’ALD. En Europe, les Pays-Bas élargissent leur programme national de dépistage néonatal de 17 à 31 maladies, dont l’ALD.
2016: Le 16 février, l’ALD est ajoutée au Panel de recommandations pour un dépistage uniforme aux États-Unis (RUSP). Aux États-Unis, California introduit le dépistage néonatal de l’ALD.
L’huile de Lorenzo
Régime restreint en AGTLC
L’anomalie biochimique de l’ALD a été identifiée pour la première fois en 1976 (Igarashi et al. 1976). Les personnes touchées avaient un niveau élevé en acides gras saturés à très longue chaîne (AGTLC). La mise en évidence de cette anomalie et le fait qu’une partie de ces acides gras étaient d’origine alimentaire a motivé les premières tentatives visant à diminuer les taux sanguins d’AGTLC par un changement de régime alimentaire. Ce régime était très limitant quant aux choix alimentaires car les AGTLC sont présents dans de nombreux produits végétaux et animaux. Malheureusement, la restriction alimentaire en acides gras C26:0 n’entraine pas la baisse des taux plasmatiques d’AGTLC chez les patients atteints d’ALD (Brown et al. 1982).
 Figure: L’ajout d’acides gras en C18 mono-insaturés :1 et C22:1 aux cellules ALD, réduit la synthèse de C26:0, parce que les enzymes qui sont responsables de sa synthèse (élongases) ont une plus grande affinité pour les acides gras mono-insaturés.
Figure: L’ajout d’acides gras en C18 mono-insaturés :1 et C22:1 aux cellules ALD, réduit la synthèse de C26:0, parce que les enzymes qui sont responsables de sa synthèse (élongases) ont une plus grande affinité pour les acides gras mono-insaturés.
Inhiber la synthèse d’AGTLC
L’absence d’effet du régime alimentaire a permis de comprendre que le corps devait aussi produire une quantité importante d’AGTLC. En effet, la majorité des AGTLC est produite à partir d’acides gras à longue chaîne (Tsuji et al. 1981). Il y a eu diverses tentatives pour interférer avec l’élongation des chaînes d’acides gras. La première approche utilisait les acides gras mono-insaturés à longue chaîne. Ces acides gras mono-insaturés à longue chaîne diffèrent des acides gras saturés à longue chaîne parce qu’ils ont dans la chaîne grasse une double liaison. L’hypothèse était que les acides gras mono-insaturés seraient en compétition avec les acides gras saturés pour se lier aux enzymes responsables de l’allongement des très longues chaînes d’acides gras (les élongases) et que ces enzymes auraient une affinité plus grande pour les acides gras mono-insaturés que pour les acides gras saturés. La conséquence étant une diminution de la production d’AGTLC saturés (Figure). En effet, l’addition d’acides gras mono-insaturés, en particulier l’acide oléique (C18:1), dans le milieu de culture de fibroblastes de peau provenant de patients ALD réduit la synthèse de C26:0 de 60% (Rizzo et al. 1984).
L’huile de Lorenzo
L’ajout d’acide oléique (C18:1) puis d’acide érucique (C22:1) utilisés en combinaison avec un régime relativement pauvre en matières grasses entraîne une diminution des AGTLC en 6 à 8 semaines chez les patients atteints d’ALD (Moser et al. 1987; Rizzo et al. 1989) (Figure 2). Cette huile, qui est un mélange 4:1 de glycéryl-tri-oléate (GTO) et glycéryl-tri-érucate (GTE) a été baptisée Huile de Lorenzo. L’huile de Lorenzo est prise par voie orale et est généralement bien tolérée même si une diminution modérée des plaquettes et une élévation des transaminases hépatiques ont été observées.

L’huile de Lorenzo réduit les taux plasmatiques d’AGTLC.
Dès les premières études examinant le rôle de ce mélange chez les patients atteints d’ALD, il était très clair qu’il n’influençait pas la progression de la maladie chez les personnes touchées par la forme cérébrale. Ce résultat a été obtenu de façon répété. L’huile de Lorenzo ne modifie pas le cours de l’ALD cérébrale chez l’enfant ou chez l’adulte et n’a jamais été indiquée dans la forme cérébrale de la maladie. En outre, son effet n’a jamais été étudié en vue d’une greffe de moelle osseuse ni dans la période suivant ce traitement.
Les premières études sur la forme adulte de l’ALD, l’adrénomyéloneuropathie (AMN), ont utilisé ce traitement sur des petites populations avec des présentations cliniques variables et sur de courtes périodes sans montrer d’effet clair. Ces essais cliniques très limités ont conduit à de grandes déclarations sur l’absence d’un effet et ont soulevé la question de l’entrée des principes actifs dans le cerveau. En effet, tandis que l’huile de Lorenzo diminue les niveaux de C26:0 dans le plasma et les tissus adipeux, elle ne modifie pas les niveaux de C26:0 dans le cerveau (Rasmussen et al 1994). Les auteurs ont conclu que : « l’échec de l’entrée dans le cerveau d’une quantité significative d’acide érucique est peut-être le facteur responsable des résultats décevants d’une thérapie diététique de l’ALD ». Ceci peut être expliqué par une autre étude qui a fourni les preuves expérimentales que l’acide érucique n’entre pas dans le cerveau et qu’il est rapidement métabolisé (Golovko and Murphy 2006).
Dans les années 1990, plusieurs groupes ont commencé à étudier indépendamment l’utilisation de l’huile de Lorenzo comme thérapie préventive – un agent qui pourrait, soit prévenir une évolution cérébrale chez les garçons à risque n’ayant pas de symptômes cérébraux, soit ralentir la progression de la maladie chez les hommes ayant une AMN. Deux essais (Aubourg et al. 1993; Van Geel et al. 1999) ont montré que l’huile de Lorenzo ne modifie pas la progression de la maladie chez les hommes ayant une AMN. De plus, quelques hommes ayant une AMN ont développé une atteinte cérébrale malgré l’utilisation de l’huile. Tous ces essais étaient des essais ouverts (l’huile de Lorenzo n’a pas été testée contre un placebo) et étaient donc difficilement irrévocable.
Souhaitant fournir des réponses définitives concernant l’efficacité clinique de l’huile de Lorenzo en tant qu’agent modificateur de l’AMN, un essai de phase III AMN a été lancé en 2007 à l’Institut Kennedy Krieger. L’étude de 4 ans incluait 120 hommes atteints d’AMN sans atteinte cérébrale, et 120 femmes ayant une ALD avec myélopathie. Les taux de progression étaient comparés dans les groupes recevant l’huile de Lorenzo et le placebo à l’aide de l’échelle EDSS Kurtzke comme résultat principal, et d’une variété de résultats secondaires. Malheureusement, l’étude a été interrompue avant la fin par le conseil de surveillance de la sécurité en raison des effets secondaires présumés du placebo. En conséquence, il n’y a pas de preuve scientifique d’un effet modificateur de l’huile de Lorenzo dans l’AMN.
L’effet de l’huile de Lorenzo sur l’ALD cérébrale a été étudié par Moser et al (Moser et al. 2005) sur un grand nombre de garçons ayant une IRM normale à leur inclusion dans l’étude. Dans ce groupe, les AGTLC diminuaient après traitement et un lien a pu être établi entre une diminution du risque de développer une maladie cérébrale et la baisse des taux d’AGTLC. Cette étude, cependant, présente plusieurs limites, comme par exemple l’utilisation de contrôles historiques au lieu d’un groupe placebo, et n’offre aucune preuve définitive de l’efficacité de l’huile de Lorenzo dans la prévention de l’apparition des ALD cérébrales chez les garçons ayant une ALD. Sur la base des résultats de cette étude, l’huile de Lorenzo est donc proposée dans certains pays aux garçons ayant une ALD dans le but de réduire le risque d’apparition d’une ALD cérébrale. Il convient de noter que l’utilisation de l’huile de Lorenzo n’est pas sans effets secondaires et a un impact considérable sur la qualité de vie.
Nous recommandons que l’huile de Lorenzo ne soit mise en place que par les centres qui ont la capacité d’assurer un suivi par IRM, de prodiguer des conseils nutritionnels, et ayant la capacité de mesurer les AGTLC et d’autres acides gras essentiels. D’autres centres considèrent que l’efficacité de l’huile de Lorenzo n’est pas prouvée et ne recommandent pas son utilisation.
La prise d’huile de Lorenzo ne doit jamais être initiée sans la surveillance d’un médecin. Bien que des effets indésirables graves n’aient pas été observés lors de son utilisation contrôlée, la sécurité des individus traités par l’huile de Lorenzo doit être évaluée régulièrement. Enfin, il faut dire clairement que l’huile de Lorenzo est indiquée uniquement pour le traitement du défaut biochimique des ALD et ne joue aucun rôle dans d’autres pathologies neurologiques démyélinisantes ou progressives dans lesquelles les personnes peuvent métaboliser les acides gras à très longue chaîne.
La lovastatine pour l’ALD
Authors: Marc Engelen, M.D., Ph.D. & Stephan Kemp, Ph.D.
Traduction Virginie Bonnamain, Ph.D.
New England Journal of Medicine 2010 Jan 21;362(3):276-277.
En 1999, il a été rapporté que la lovastatine, un médicament utilisé pour baisser le taux de cholestérol, réduisait les acides gras à très longue chaîne (AGTLC) dans le sang de patients présentant une adrénoleucodystrophie liée à l’X (Singh et al. 1998). À la suite de cette publication, de nombreux patients ALD dans le monde entier ont commencé à utiliser la lovastatine dans l’espoir que cette molécule ait un effet positif sur l’évolution clinique de leur maladie. Au cours des années suivantes, d’autres chercheurs ont mis en doute l’effet bénéfique de la lovastatine sur les AGTLC. Une étude ultérieure portant sur la simvastatine (une statine structurellement apparentée) chez des garçons atteints d’ALD cérébrale n’a montré aucun effet sur les AGTLC (Verrips et al. 2000). Et le traitement des souris ALD n’a eu aucun effet sur les taux d’AGTLC dans le cerveau et les glandes surrénales (Cartier et al. 2000, Yamada et al. 2000).
Afin de déterminer si la lovastatine pouvait réellement réduire les taux d’AGTLC plasmatiques chez les patients atteints d’ALD, un essai croisé randomisé en double aveugle contre placebo a été mené (Figure). Quatorze patients présentant le phénotype adrénomyéloneuropathie (AMN) ont participé à l’essai. Les patients ont été traités avec la lovastatine pendant 6 mois puis avec un placebo pendant 6 mois, ou inversement. Ni les patients ni les chercheurs ne savaient qui utilisait quel type de médicament pendant la période de traitement. À la fin de l’essai, les échantillons et les données ont été analysés.
Comme prévu, le traitement par la lovastatine a diminué les taux plasmatiques de cholestérol LDL. Les taux plasmatiques d’AGTLC ont diminué d’environ 20%, mais ils sont restés 2 à 3 fois plus élevés que la normale. De plus, les taux d’AGTLC dans les globules rouges et blancs sont restés inchangés après le traitement par la lovastatine. Étant donné que les AGTLC sont pratiquement insolubles dans l’eau, la plupart des AGTLC dans le sang sont transportés sous forme d’esters de cholestérol dans des particules de lipoprotéines telles que les LDL. Lorsque les particules de LDL ont été analysées, aucun effet sur les niveaux d’AGTLC dans ces particules n’a été observé.
Les auteurs ont conclu que la lovastatine entraînait une légère diminution du taux plasmatique d’AGTLC, ce qui doit être considéré comme une conséquence non spécifique de la diminution du LDL-cholestérol. Les médecins ne doivent pas prescrire la lovastatine aux patients atteints d’ALD comme traitement pour diminuer les taux d’AGTLC, car les données des études effectuées ne le justifient pas.  Figure: Essai croisé randomisé, à double aveugle, contre placebo, visant à tester l’effet de la lovastatine sur les taux d’AGTLC dans le plasma et les cellules sanguines de patients atteints d’ALD.
Figure: Essai croisé randomisé, à double aveugle, contre placebo, visant à tester l’effet de la lovastatine sur les taux d’AGTLC dans le plasma et les cellules sanguines de patients atteints d’ALD.
L’insuffisance surrénale
Authors: Gabor Linthorst, M.D., Ph.D. and Stephan Kemp, Ph.D.
Traduction Virginie Bonnamain, Ph.D.
Hommes
 L’insuffisance surrénale (ou maladie d’Addison) est souvent observée dans les cas d’ALD:
L’insuffisance surrénale (ou maladie d’Addison) est souvent observée dans les cas d’ALD:
- La grande majorité (80% ou plus) des patients atteints d’ALD cérébrale infantile et d’adrénomyéloneuropathie (AMN) présentent des signes cliniques et biochimiques d’insuffisance surrénale. Dans ce groupe de patients, l’insuffisance surrénale accompagne les autres symptômes (neurologiques) de l’ALD. Cependant, il arrive que certains patients ALD ne développent pas d’insuffisance surrénale, malgré une atteinte neurologique sévère (même jusqu’à un âge avancé).
- Chez certains autres patients ALD, l’insuffisance surrénale peut être le seul symptôme, ce sous-groupe est de ce fait surnommé le «phénotype Addison seul».
Bien que l’insuffisance surrénale précède souvent l’apparition des symptômes neurologiques, aucune caractéristique de la maladie (par exemple, le génotype de l’ALD ou les concentrations en AGTLC) ne permet de prédire avec précision l’apparition d’une insuffisance surrénale chez un patient ALD donné.
Une insuffisance surrénale non reconnue peut souvent entraîner une morbidité (conséquences ou complications [autres que le décès] résultant de la maladie) et même une mortalité. Des évaluations périodiques minutieuses peuvent identifier une réserve surrénale altérée à un stade précoce et faciliter l’initiation du traitement.
L’insuffisance surrénale entraîne une diminution de la production de cortisol, l’hormone du stress, par la glande surrénale. Une diminution graduelle des niveaux de cortisol passe souvent inaperçue. Les symptômes peuvent inclure fatigue, hypotension orthostatique (tension artérielle basse en position debout ou en se levant), perte de poids, brunissement de la peau et des gencives. Cependant, une augmentation du taux de cortisol est nécessaire lorsque le corps est confronté à un stress physique, tel qu’une infection virale (rhume) ou pire. Dans de tels cas, le manque de cortisol peut entraîner une hypotension dangereuse (également appelée crise addisonienne).
La présentation d’une crise addisonienne a été rapportée comme la première présentation de l’ALD et peut également nécessiter une intervention médicale (brève) chez les patients présentant une ALD et une insuffisance surrénale.
Une évaluation prospective de la fonction surrénale chez une cohorte de 49 garçons atteints d’ALD et neurologiquement pré-symptomatiques (âge moyen 4 ans) a montré que 80% avaient déjà une insuffisance de la fonction surrénale (Dubey et al. 2005). Par conséquent, les endocrinologues doivent rechercher les AGTLC chez les garçons et les hommes atteints d’insuffisance surrénale lorsque les tests d’auto-anticorps dirigés contre le cortex surrénal sont négatifs ou que des signes de myélopathie sont présents.
L’hormonothérapie est obligatoire pour tous les patients ALD souffrant d’insuffisance surrénale et peut sauver la vie. Rien n’indique que l’hormonothérapie surrénale modifie la progression neurologique due à l’ALD.
Comme l’insuffisance surrénale due à l’ALD est causée par les effets toxiques directs des AGTLC sur la glande surrénale, l’ACTH plasmatique (hormone adrénocorticotrope, nécessaire au bon fonctionnement des glandes surrénales et à la réaction du corps au stress) peut être utilisée comme marqueur de laboratoire pour détecter le dysfonctionnement surrénal chez les enfants, les adolescents et les adultes. Les tests Short Synthetix ACTH (Synacthen-test) peuvent être utilisés pour étudier la réserve surrénale en détail.
Femmes
Contrairement aux hommes affectés, l’insuffisance surrénale clinique est exceptionnellement rare chez les femmes atteintes d’ALD. Des études menées auprès d’un grand groupe de femmes atteintes d’ALD ont montré que l’insuffisance surrénale était présente chez environ 1% des femmes atteintes d’ALD.