
Trasplante de células madre hematopoyéticas
Authors: Charles Peters, M.D., Virginie Bonnamain, Ph.D. and Stephan Kemp, Ph.D.
(Traducido por Nerea Montedeoca Vázquez (Biomedicine student))
¿Qué es el trasplante de células madre hematopoyéticas?
![]() El trasplante de células madre hematopoyéticas (TCMH, también conocido como trasplante de médula ósea), es un procedimiento médico aplicado en el tratamiento de pacientes afectos de cáncer (de células sanguíneas) o ciertos tipos de enfermedades genéticas graves (como la adrenoleucodistrofia). Durante este procedimiento, el paciente recibe células madre sanas de un donante. El procedimiento se compone de 3 fases principales:
El trasplante de células madre hematopoyéticas (TCMH, también conocido como trasplante de médula ósea), es un procedimiento médico aplicado en el tratamiento de pacientes afectos de cáncer (de células sanguíneas) o ciertos tipos de enfermedades genéticas graves (como la adrenoleucodistrofia). Durante este procedimiento, el paciente recibe células madre sanas de un donante. El procedimiento se compone de 3 fases principales:
- La extracción de células madre hematopoyéticas de la sangre. Esto puede realizarse utilizando las células del propio paciente o las de un donante compatible o similar genéticamente. Este procedimiento se denomina “aféresis” o “leucaféresis”.
- Quimioterapia para eliminar la médula ósea del paciente y evitar que esta siga produciendo más células madre “enfermas”. Este procedimiento se denomina “condicionamiento”. El condicionamiento también permite crear espacio en la médula ósea para las células madre que serán trasplantadas
- La infusión intravenosa de células madre hematopoyéticas, también denominada trasplante.
¿Qué es una célula madre hematopoyética?
Una célula madre es un tipo de células que tiene la habilidad única de producir tipos celulares especializados en el cuerpo (como las células cardíacas, las células de la piel, las células cerebrales, etc.).
Una célula madre hematopoyética es una célula madre que puede dar lugar específicamente a diferentes células sanguíneas, tales como los glóbulos rojos, los glóbulos blancos y las plaquetas: así como a todas las células del sistema inmune. Las células madre hematopoyéticas se localizan principalmente en la médula ósea, que es un tejido con una estructura similar a una esponja el cuál se encuentra en el interior de los huesos grandes del cuerpo. Sin embargo, un pequeño número de estas células se halla también en la sangre.
Antes de la extracción de las células madre hematopoyéticas, el donante recibe un fármaco que induce un incremento en la liberación de células madre hematopoyéticas de la médula ósea a la sangre. Las células madre hematopoyéticas pueden entonces ser extraídas directamente del torrente sanguíneo. Esto evita tener que realizar el procedimiento de aspirado de médula ósea, que resulta mucho más complejo.
Existen dos tipos de trasplante de células madre hematopoyéticas: el alogénico y el autólogo
En el caso de un trasplante alogénico de células madre hematopoyéticas, estas células proceden de un donante compatible o genéticamente similar. Esta persona puede ser un pariente (un hermano o hermana en condiciones ideales) o, cuando un hermano compatible no se halle disponible, una persona no relacionada con el paciente. En determinadas condiciones, la sangre procedente del cordón umbilical podría ser también utilizada como una fuente de células madre hematopoyéticas.
En este entorno alogénico, se deben tomar medidas para minimizar o eliminar una posible complicación fatal en la cual las células inmunitarias derivadas del donante podrían atacar las células y tejidos del receptor. Esta reacción recibe el nombre de enfermedad de injerto contra huésped (EICH). Además, una supresión suficiente del sistema inmune del receptor debe ser alcanzada con tal de reducir la probabilidad de que el sistema inmune del receptor ataque y rechace a las células del donante.
En el caso de un trasplante autólogo de células madre hematopoyéticas, las propias células madre hematopoyéticas del paciente son extraídas antes del proceso de condicionamiento y trasplantadas de vuelta al paciente tras, por ejemplo, una corrección mediante terapia génica.
Vídeo explicativo sobre los conceptos básicos en el trasplante de células madre
https://www.youtube.com/watch?feature=player_embedded&v=7KzCc90z1Ts
Producido por bluebird bio, Inc.
¿Cómo funciona el trasplante de células madre hematopoyéticas?
Las células madre hematopoyéticas trasplantadas del donante viajan a la médula ósea, lugar donde se establecen y empiezan a generar células sanguíneas. Tras un período de varias semanas a meses, las células madre se habrán establecido y habrán producido suficientes células para repoblar el ya “extirpado” sistema productor sanguíneo del paciente. Sin embargo, la recuperación del sistema inmune requiere un período prolongado y, durante este tiempo, el paciente trasplantado corre el riesgo de desarrollar una variedad de infecciones “oportunistas” que podrían suponer una amenaza potencial para la vida del paciente.
Durante las pasadas 4 décadas, el trasplante de células madre hematopoyéticas ha sido utilizado exitosamente para tratar una amplia variedad de enfermedades, incluyendo la leucemia, el linfoma, los tumores sólidos malignos, la anemia aplásica y las enfermedades relacionadas con el fallo de médula ósea, las enfermedades relacionadas con inmunodeficiencia, hemoglobinopatías como la anemia falciforme y la talasemia, así como enfermedades genéticas como por ejemplo las enfermedades de almacenamiento lisosomal y múltiples leucodistrofias como la adrenoleucodistrofia.
¿Cómo funciona el trasplante de células madre hematopoyéticas para el tratamiento de la adrenoleucodistrofia cerebral?
En el caso de las enfermedades genéticas, el trasplante alogénico de células madre hematopoyéticas representa una forma de terapia génica “adoptiva”. Las células del donante son capaces de producir la enzima/proteína que no es funcional en las propias células del paciente. En el caso de la adrenoleucodistrofia cerebral, este mecanismo y/o la corrección de la destrucción de la mielina mediada por el sistema inmune pueden ser la razón para su efectividad. Sin embargo, el trasplante alogénico de células madre hematopoyéticas para la adrenoleucodistrofia cerebral presenta algunos desafíos:
- Existe un riesgo de desarrollar la enfermedad de injerto contra huésped tras el trasplante de células madre hematopoyéticas, la cuál ha sido asociada con una mayor progresión o agravamiento de la enfermedad cerebral.
- Además, con tal de que se produzca una protección y un beneficio que se mantenga en el tiempo derivados del trasplante de células madre hematopoyéticas, los chicos con adrenoleucodistrofia cerebral deben continuar presentando un grado significativo de células derivadas del donante.
Se debe tener en cuenta que la potenciación de gadolinio observada en las resonancias magnéticas cerebrales de chicos con adrenoleucodistrofia cerebral, se encuentra asociada a la desmielinización “activa”. La desaparición de esta potenciación ha sido observada en un mes como período más temprano después del trasplante de células madre hematopoyéticas en el entorno de un injerto procedente de un donante. No obstante, el tiempo requerido para que un trasplante exitoso de células madre hematopoyéticas derivadas de un donante detenga de forma efectiva la progresión de la desmielinización en la adrenoleucodistrofia cerebral se mide en meses, normalmente de 6 a 12.
Resumen histórico de la experiencia con trasplantes alogénicos de células madre hematopoyéticas en el tratamiento de la adrenoleucodistrofia cerebral
1982: El primer trasplante de médula ósea para la adrenoleucodistrofia cerebral infantil se realiza en un niño con un estadio avanzado de la enfermedad (es decir, muy tarde). El paciente murió 141 días tras el trasplante debido a la progresión de la enfermedad, a pesar de haber tenido un trasplante exitoso (las células madre hematopoyéticas del donante viajaron hasta la médula ósea y comenzaron a producir células sanguíneas de forma exitosa) (Moser et al., 1984).
1990: Aubourg y colegas en París describieron el primer trasplante de médula ósea exitoso en un niño con un estadio muy temprano de la enfermedad cerebral (Aubourg et al., 1990).
2000: Shapiro y colegas describieron resultados a largo plazo (es decir, 5-10 años) del trasplante alogénico de células madre hematopoyéticas en 12 niños con adrenoleucodistrofia cerebral, lo cual mostró perspectivas para alcanzar estabilidad en la desmielinización y cambios en la función neuropsicológica (Shapiro et al., 2000).
2004: La experiencia internacional en los trasplante de células madre hematopoyéticas de 1982 a 1999 para la adrenoleucodistrofia cerebral infantil y adolescente fue recogida a través de un consorcio de 43 centros especializados en el trasplante de células madre hematopoyéticas (Peters et al., 2004). Este fue el primer informe exhaustivo de la experiencia mundial con el TCMH para esta enfermedad, el cual describía los resultados de 126 pacientes.
2007: Mahmood y colegas analizaron las probabilidades de supervivencia en niños con adrenoleucodistrofia cerebral los cuáles no habían recibido un trasplante de células madre hematopoyéticas (Mahmood et al., 2007). En un subgrupo de estos pacientes con enfermedad cerebral temprana, compararon la supervivencia en aquellos niños que recibieron un trasplante de células madre hematopoyéticas con aquellos niños que no lo recibieron. La probabilidad de supervivencia a los 5 años en el grupo de no trasplantados resultó significativamente más baja que la probabilidad de supervivencia a los 5 años en el 95% del grupo de trasplantados con un estadio temprano de enfermedad cerebral. Los autores concluyeron que el trasplante de células madre hematopoyéticas en los estadios tempranos y progresivos de la adrenoleucodistrofia cerebral infantil es beneficioso. Recomendaron que el trasplante de células madre hematopoyéticas debería ser ofrecido a chicos en estadios tempranos de la enfermedad cerebral.
1997-2011: Se emitieron numerosos informes de instituciones describiendo su experiencia con el trasplante de células madre hematopoyéticas, el uso de condicionamiento de intensidad reducida y la sangre procedente del cordón umbilical como fuente de células madre sanguíneas.
2015: Van Geel y colegas investigaron si los pacientes con adrenoleucodistrofia que recibieron un trasplante de células madre hematopoyéticas para la adrenoleucodistrofia cerebral en la infancia aún podrían desarrollar la enfermedad de la médula espinal con aparición durante la edad adulta (adrenomieloneuropatía, AMN). Este estudio observacional retrospectivo encontró que tres de cada cinco pacientes que recibieron un trasplante de células madre hematopoyéticas durante la infancia desarrollaron signos de mielopatía en la edad adulta. Estos datos sugieren que el trasplante de células madre hematopoyéticas para la adrenoleucodistrofia cerebral en la infancia no parece prevenir la aparición de mielopatía y neuropatía periférica en la edad adulta. Estos hallazgos deberán ser confirmados en estudios independientes. No obstante, en caso de ser cierto, esto representaría consecuencias de cara al seguimiento de los pacientes que fueron trasplantados en la infancia y que están llegando a la edad adulta en la actualidad (Van Geel et al. 2015).
Experiencia con el trasplante alogénico de células madre hematopoyéticas para la adrenoleucodistrofia cerebral y lecciones aprendidas
Un aspecto importante con respecto al futuro de la terapia para la adrenoleucodistrofia cerebral y específicamente para el trasplante de células madre hematopoyéticas es el desafío de tratar a niños con un estadio avanzado de la enfermedad. Con tal de tratar a niños en un estadio avanzado de la enfermedad cerebral (es decir, puntuación de severidad en IRM ≥9) de forma más efectiva con el trasplante de células madre hematopoyéticas es imperativo tratar los siguientes puntos: (1) desarrollar un método para evaluar de forma precisa la rapidez de la enfermedad cerebral; (2) explorar más exhaustivamente el uso de la N-acetilcisteína; (3) evaluar el uso de los regímenes de condicionamiento de intensidad reducida en los trasplantes de células madre hematopoyéticas, (4) investigar terapias combinadas que incluyan células madre complementarias o alternativas como por ejemplo las células madre neurales; (5) desarrollar e implementar maneras más efectivas de detener rápidamente la desmielinización cerebral, reparar la mielina dañada y restaurar la funcionalidad perdida.
La supervivencia, de excelente a sobresaliente generalmente, y los resultados funcionales observados tras el trasplante de células madre hematopoyéticas en niños con un estadio temprano de la enfermedad cerebral (es decir, puntuación de severidad en IRM <9 y ≤3 en particular) estableció las bases para implementar el cribado neonatal para la adrenoleucodistrofia (véase Cribado Neonatal). Además, sería altamente beneficioso y de extrema importancia identificar marcadores que predigan qué niños con adrenoleucodistrofia están destinados a desarrollar adrenoleucodistrofia cerebral y, en consecuencia, requerirán de un trasplante de células madre hematopoyéticas.
Es importante tener en cuenta que actualmente no existe ningún tipo de indicación para la utilización del trasplante de células madre hematopoyéticas en hombres y mujeres adultos con la forma de la enfermedad que afecta a la médula espinal, tanto con como sin síntomas.
Mientras que se han realizado avances significativos en la reducción de riesgos asociados al trasplante de células madre hematopoyéticas, particularmente, en aquellos procedentes de donantes no emparentados de médula ósea o sangre de cordón umbilical; aun existe un riesgo significativo de mortalidad asociada al trasplante, además de riesgo de muerte debido a la progresión de la enfermedad. Con los avances realizados en terapia génica (Véase la página Terapia génica para la ALD) una alternativa al trasplante de células madre hematopoyéticas debería ser considerada en casos específicos (por ejemplo, ante la falta de disponibilidad de un donante compatible adecuado de células madre sanguíneas).
Terapia génica para la ALD
Authors: Virginie Bonnamain, Ph.D. and Stephan Kemp, Ph.D.
(Traducido por Nerea Montedeoca Vázquez (Biomedicine student))
En 2009, los Dres. Nathalie Cartier, Patrick Aubourg y colegas (Hospital Saint Vincent de Paul, París, Francia) informó del exitoso tratamiento de dos niños de 7 años con signos tempranos de adrenoleucodistrofia cerebral mediante terapia génica (Cartier et al. Science). Estos niños fueron candidatos para el trasplante alogénico de células madre hematopoyéticas (TCMH), pero no se encontraron donantes compatibles.
La terapia génica se basa en corregir las células de la médula ósea de los propios pacientes:
- Las células de la médula ósea fueron extraídas de los pacientes. Este proceso se denomina “aféresis”.
- En el laboratorio, una copia normal del gen de la adrenoleucodistrofia fue insertada en el interior de las células de la médula ósea mediante la utilización de un virus derivado del VIH.
- Los pacientes fueron sometidos a quimioterapia con el objetivo de eliminar su propia médula ósea para evitar que esta siguiese produciendo más células madre. Este procedimiento se denomina “condicionamiento”.
- A continuación, se les reinfundieron sus propias células genéticamente corregidas, las cuáles presentaban el gen normal para la adrenoleucodistrofia.
Vídeo explicativo sobre los conceptos básicos en el trasplante de células madre
https://www.youtube.com/watch?feature=player_embedded&v=7KzCc90z1Ts
Producido por bluebird bio, Inc.
Los dos pacientes fueron evaluados durante 24 y 30 meses. A lo largo de este período, el 15% de sus células sanguíneas mostraron expresión de la proteína de la adrenoleucodistrofia normal, mientras que antes de la terapia, no se detectó nada de proteína de la adrenoleucodistrofia normal en las células sanguíneas de los pacientes. Los VLCFA fueron reducidos alrededor de un 38% en el plasma de los pacientes.
Los escáneres cerebrales de RM y las pruebas cognitivas mostraron que la progresión de la enfermedad cerebral se detuvo pasados los 14-16 meses. Los pacientes permanecieron estables desde entonces. La lesión desmielinizante observada en la vía auditiva de uno de los pacientes fue revertida. La detención de la desmielinización cerebral progresiva en estos pacientes representa un resultado clínico comparable al obtenido mediante el TCMH.
Estudio Starbeam
El Estudio Starbeam (ALD-102, NCT01896102) es un estudio experimental en fase 2/3 sobre la terapia génica. Los objetivos consisten en determinar la seguridad y tolerabilidad del tratamiento de administración única mediante terapia génica conocido como “Lenti-D”, y en determinar si el tratamiento puede detener la progresión de la adrenoleucodistrofia cerebral. Desde el mes de abril de 2018, un total de 31 pacientes fueron incluidos en el estudio. De estos 31 pacientes, 29 fueron tratados con Lenti-D y la mediana en cuanto al seguimiento para todos los pacientes tratados fue de 34 meses (0,4 – 54 meses).
El principal criterio de valoración respecto de la eficacia para el estudio Starbeam es la ausencia de Discapacidades Funcionales Mayores (Major Functional Disabilities, MFDs) a los 24 meses tras el trasplante. Las MFDs se corresponden con discapacidades graves, las cuales se cree que puedan tener un gran impacto en la habilidad del paciente para valerse de forma independiente: pérdida de la capacidad para comunicarse, ceguera cortical, necesidad de ser alimentado mediante sondas, incontinencia total, dependencia de la silla de ruedas y pérdida completa del movimiento voluntario. El segundo criterio de valoración del estudio incluye la progresión de la enfermedad cerebral. Esto es evaluado mediante la intensificación de gadolinio en la RM cerebral (que es un indicador de neuroinflamación) y a través de la Puntuación de la Función Neurológica (Neurologic Function Score, NFS). La NFS se trata de un sistema de puntuación utilizado para evaluar la severidad de los déficits clínicos al calificar 15 síntomas en múltiples dominios). También se evaluó el perfil de seguridad de Lenti-D (efectos secundarios y análisis de la integración del gen en el genoma).
En octubre de 2017, los resultados de 17 pacientes tratados con Lenti-D, los cuales completaron un seguimiento durante 24 meses, fueron publicados (Eichler et al. 2017). En el momento del análisis de los datos para este grupo inicial de 17 pacientes, la mediana para el seguimiento era de 29,4 meses (21,6-42 meses).
- 15 (88%) de 17 pacientes sobrevivieron, con síntomas clínicos mínimos.
- 1 paciente murió debido a la progresión de la enfermedad durante el condicionamiento pre-trasplante.
- 1 paciente fue retirado del estudio y falleció debido a complicaciones derivadas de un trasplante alogénico posterior.
- De los 15 pacientes restantes, no se observaron indicios de la enfermedad del injerto contra el huésped (que se produce cuando las células trasplantadas “atacan” a las células del paciente).
- 14 de estos 15 pacientes obtuvieron una puntuación de entre 0 y 1 en la NFS, lo cual indica ausencia o síntomas clínicos mínimos.
- 12 de los 15 pacientes obtuvieron una puntuación estable en la escala Loes, lo cual indica ausencia de progresión de la lesión.
- La intensificación de gadolinio, que se encontraba presente desde el inicio en todos los pacientes, resultó hallarse ausente en los 15 pacientes alrededor del sexto mes postrasplante. En unos pocos pacientes, se produjo una reemergencia de la intensificación de gadolinio en varios momentos (incluyendo a 2 pacientes en el mes 24), pero esta potenciación resultó ser menos extensa que la intensificación de gadolinio que se hallaba presente desde el inicio.
Estos primeros resultados sugieren que el tratamiento de la adrenoleucodistrofia cerebral mediante terapia génica con Lenti-D es como mínimo igual de efectivo que el trasplante alogénico convencional. La ausencia de la enfermedad del injerto contra el huésped indican que posiblemente esta terapia sea más segura
Tras completar el estudio ALD-102 (aproximadamente 2 años), los pacientes fueron incluidos en un nuevo estudio (LTF-304, NCT02698579) para seguir a los pacientes durante 13 años más y evaluar así la seguridad y eficacia de la terapia a largo plazo.
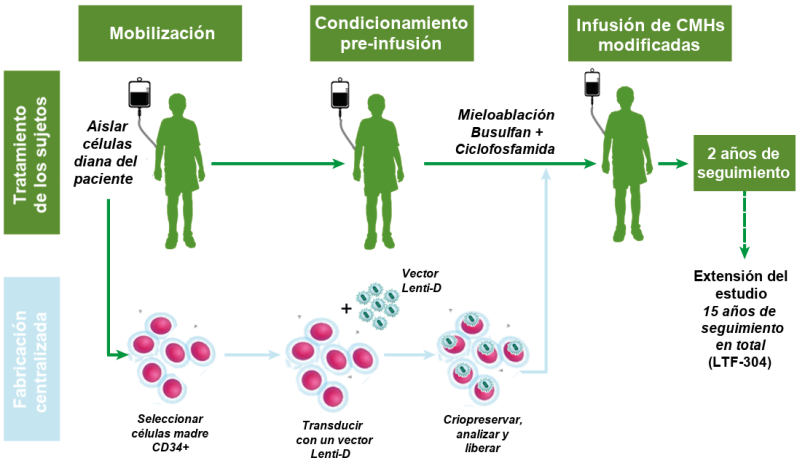 Figura: Resumen del protocolo para el tratamiento del estudio Starbeam. Imagen cedida por cortesía de bluebird bio.
Figura: Resumen del protocolo para el tratamiento del estudio Starbeam. Imagen cedida por cortesía de bluebird bio.
El patrocinador del estudio Starbeam, bluebird bio, es una compañía biotecnológica localizada en Cambridge, MA, EE.UU.
Datos sobre la ALD
Authors: Marc Engelen, M.D., Ph.D., Rachel Salzman, D.V.M. (CSO, The Stop ALD Foundation) & Stephan Kemp, Ph.D.
(Traducido por Nerea Montedeoca Vázquez (Biomedicine student))
Definición
La adrenoleucodistrofia ligada al cromosoma X (ALD) es un grave trastorno genético y progresivo que afecta a las glándulas suprarrenales, la médula espinal y la sustancia blanca (mielina) del sistema nervioso. Se describió por primera vez en 1923 y ha sido conocida como la enfermedad de Schilder y leucodistrofia sudanofílica. En 1970, se introdujo el nombre adrenoleucodistrofia, con el objetivo de describir de forma más fidedigna las manifestaciones de la enfermedad. ‘Adreno’ se refiere a las glándulas suprarrenales; ‘Leuco’ se refiere a la sustancia blanca del cerebro, y ‘distrofia’ significa crecimiento o desarrollo anormal. Esta enfermedad no presenta ninguna relación con “la adrenoleucodistrofia neonatal”, que pertenece a las enfermedades de la biogénesis peroxisomal del espectro Zellweger.
Bioquímica
La adrenoleucodistrofia es una enfermedad hereditaria de almacenamiento metabólico en la cual un defecto en una enzima específica resulta en la acumulación de ácidos grasos de cadena muy larga (Very long chain fatty acids, VLCFA) en todos los tejidos del cuerpo. Estos VLCFA son perjudiciales para algunas células y órganos. Por razones que aún no han sido resueltas, el cerebro, la médula espinal, los testículos y las glándulas suprarrenales se ven afectados principalmente. En el sistema nervioso central la acumulación de VLCFA finalmente destruye la vaina de mielina que rodea los nervios causando problemas neurológicos. Los VLCFA son tóxicos para las células de la glándula suprarrenal y su mal funcionamiento causa la enfermedad de Addison (insuficiencia suprarrenal).
 Figura 1: los VLCFA que se acumulan en la adrenoleucodistrofia son el resultado, mayoritariamente, de la elongación de los ácidos grasos de cadena larga. Para mantener el equilibrio justo en la homeostasis de los VLCFA, su exceso ha de ser degradado. Los VLCFA sólo pueden ser degradados en los peroxisomas. Todas las células del cuerpo, a excepción de los glóbulos rojos, tienen peroxisomas. La Adrenoleucodistrofia es causada por mutaciones en el gen ABCD1 que produce la proteína de la adrenoleucodistrofia (ALDP). La ALDP actúa como un transportador de VLCFA desde el citosol hasta el interior del peroxisoma. Una deficiencia de ALDP bloquea este transporte, lo que resulta en la degradación alterada de estos ácidos grasos y en una posterior acumulación de estos en células, tejidos y órganos. Las enzimas necesarias para la degradación de los VLCFA están presentes dentro de los peroxisomas, pero estos no pueden llegar hasta estas enzimas.
Figura 1: los VLCFA que se acumulan en la adrenoleucodistrofia son el resultado, mayoritariamente, de la elongación de los ácidos grasos de cadena larga. Para mantener el equilibrio justo en la homeostasis de los VLCFA, su exceso ha de ser degradado. Los VLCFA sólo pueden ser degradados en los peroxisomas. Todas las células del cuerpo, a excepción de los glóbulos rojos, tienen peroxisomas. La Adrenoleucodistrofia es causada por mutaciones en el gen ABCD1 que produce la proteína de la adrenoleucodistrofia (ALDP). La ALDP actúa como un transportador de VLCFA desde el citosol hasta el interior del peroxisoma. Una deficiencia de ALDP bloquea este transporte, lo que resulta en la degradación alterada de estos ácidos grasos y en una posterior acumulación de estos en células, tejidos y órganos. Las enzimas necesarias para la degradación de los VLCFA están presentes dentro de los peroxisomas, pero estos no pueden llegar hasta estas enzimas.
Epidemiologia
La adrenoleucodistrofia se produce en todo el mundo y no se limita a determinados grupos étnicos. La incidencia global de adrenoleucodistrofia es aproximadamente de 1 de cada 15.000 recién nacidos.
Genética
La adrenoleucodistrofia es una enfermedad ligada al cromosoma X, lo que significa que el gen de la adrenoleucodistrofia (ABCD1) se encuentra en el cromosoma X. Los hombres tienen un cromosoma X y un cromosoma (XY; Figura 2). Cuando el padre es portador del gen de la adrenoleucodistrofia afectado, no hay ningún otro cromosoma X que le otorgue protección; por lo tanto, experimentará síntomas de la adrenoleucodistrofia. Las mujeres tienen dos cromosomas X (XX; Figura 2). Las mujeres que presentan el gen defectuoso solían ser denominadas portadoras. En el pasado se creía que sólo un pequeño porcentaje de estas mujeres desarrollaría síntomas clínicos. Ahora está claro que eso no es verdad véase más abajo y la página de presentación clínica (Females with ALD). Los síntomas clínicos en las mujeres son algo más suaves que en hombres. Sin embargo, el 80% de las mujeres con adrenoleucodistrofia desarrollan síntomas clínicos. En consecuencia, las mujeres portadoras deben ser consideradas como pacientes de adrenoleucodistrofia (mujeres con adrenoleucodistrofia). La explicación más probable para que las mujeres con adrenoleucodistrofia desarrollen una forma más leve de la enfermedad es la presencia de una copia normal del gen ABCD1 en su otro cromosoma X. En las mujeres, en cada celda de uno de los cromosomas X se inactiva. Este es un proceso aleatorio a través de todas las células del cuerpo. Se cree que la presencia en los tejidos y órganos de las células que expresan la copia sana del gen ABCD1 protege a las mujeres con adrenoleucodistrofia de desarrollar la variante cerebral (adrenoleucodistrofia cerebral).
 Figura 2: (Izquierda) Si una mujer es portadora del gen defectuoso para la adrenoleucodistrofia tiene los siguientes posibles resultados después de cada embarazo: cuando el recién nacido es una niña, existe una probabilidad del 50% de que la hija reciba el gen defectuoso para la adrenoleucodistrofia y un 50% de probabilidad de que la hija no se vea afectada. En caso de que el bebé sea un niño, hay un 50% de probabilidad de que el hijo tenga adrenoleucodistrofia y un 50% de probabilidad de no verse afectado. (Derecha) Para una enfermedad ligada al cromosoma X, como la adrenoleucodistrofia, si un hombre afectado tiene hijos, entonces ninguno de sus hijos varones se verá afectado por la enfermedad (el padre siempre transmite su cromosoma Y a sus hijos). No obstante, todas sus hijas heredaran el gen defectuoso para la adrenoleucodistrofia (el padre siempre pasa su único cromosoma X (afectado) a sus hijas).
Figura 2: (Izquierda) Si una mujer es portadora del gen defectuoso para la adrenoleucodistrofia tiene los siguientes posibles resultados después de cada embarazo: cuando el recién nacido es una niña, existe una probabilidad del 50% de que la hija reciba el gen defectuoso para la adrenoleucodistrofia y un 50% de probabilidad de que la hija no se vea afectada. En caso de que el bebé sea un niño, hay un 50% de probabilidad de que el hijo tenga adrenoleucodistrofia y un 50% de probabilidad de no verse afectado. (Derecha) Para una enfermedad ligada al cromosoma X, como la adrenoleucodistrofia, si un hombre afectado tiene hijos, entonces ninguno de sus hijos varones se verá afectado por la enfermedad (el padre siempre transmite su cromosoma Y a sus hijos). No obstante, todas sus hijas heredaran el gen defectuoso para la adrenoleucodistrofia (el padre siempre pasa su único cromosoma X (afectado) a sus hijas).
Desarrollo Clínico
Los pacientes con adrenoleucodistrofia son pre-sintomáticos en el momento del nacimiento. En varones, la primera manifestación de adrenoleucodistrofia es normalmente la insuficiencia suprarrenal, que puede aparecer en bebés de pocos meses. En la edad adulta, los hombres desarrollan mielopatía (enfermedad de la médula espinal). Los varones con adrenoleucodistrofia pueden desarrollar una desmielinización cerebral progresiva (adrenoleucodistrofia cerebral), tanto en la infancia como en la edad adulta. La adrenoleucodistrofia cerebral puede tratarse de la primera manifestación de adrenoleucodistrofia o aparecer en pacientes con insuficiencia suprarrenal i/o mielopatía de base (Figura 3). Las mujeres con adrenoleucodistrofia también se encuentran afectadas y no son meras portadoras del gen afectado para la adrenoleucodistrofia, puesto que más del 80% de estas mujeres desarrollan signos y síntomas asociados a mielopatía sobre la edad de 60 años. Las mujeres con adrenoleucodistrofia raramente desarrollan insuficiencia suprarrenal o desmielinización cerebral.
 Figura 3: El espectro clínico de la adrenoleucodistrofia en hombres. Los pacientes con adrenoleucodistrofia no presentan ningún síntoma en el nacimiento. Las barras de colores muestran los rangos de edad en la aparición de la insuficiencia suprarrenal (barra azul), mielopatía (barra malva) y adrenoleucodistrofia cerebral (barra verde). La aparición de insuficiencia suprarrenal puede llegar a darse en edades tan tempranas como los 5 meses de edad. En la edad adulta, los hombres desarrollan una mielopatía progresiva crónica invariablemente. La adrenoleucodistrofia cerebral puede aparecer a cualquier edad. El paciente más joven fue diagnosticado a los 3 años de edad. El defecto primario en el gen de la adrenoleucodistrofia y el almacenamiento de VLCFA en los tejidos da lugar a la insuficiencia suprarrenal y la mielopatía (referidas en conjunto como adrenomieloneuropatía). El inicio de la adrenoleucodistrofia cerebral es más probablemente definida por la interacción del defecto primario en el gen de la adrenoleucodistrofia y una combinación de desencadenantes ambientales y/o factores genéticos (hasta el momento desconocidos). Es importante reconocer que los pacientes con insuficiencia suprarrenal y/o mielopatía permanecen en riesgo de desarrollar adrenoleucodistrofia cerebral.
Figura 3: El espectro clínico de la adrenoleucodistrofia en hombres. Los pacientes con adrenoleucodistrofia no presentan ningún síntoma en el nacimiento. Las barras de colores muestran los rangos de edad en la aparición de la insuficiencia suprarrenal (barra azul), mielopatía (barra malva) y adrenoleucodistrofia cerebral (barra verde). La aparición de insuficiencia suprarrenal puede llegar a darse en edades tan tempranas como los 5 meses de edad. En la edad adulta, los hombres desarrollan una mielopatía progresiva crónica invariablemente. La adrenoleucodistrofia cerebral puede aparecer a cualquier edad. El paciente más joven fue diagnosticado a los 3 años de edad. El defecto primario en el gen de la adrenoleucodistrofia y el almacenamiento de VLCFA en los tejidos da lugar a la insuficiencia suprarrenal y la mielopatía (referidas en conjunto como adrenomieloneuropatía). El inicio de la adrenoleucodistrofia cerebral es más probablemente definida por la interacción del defecto primario en el gen de la adrenoleucodistrofia y una combinación de desencadenantes ambientales y/o factores genéticos (hasta el momento desconocidos). Es importante reconocer que los pacientes con insuficiencia suprarrenal y/o mielopatía permanecen en riesgo de desarrollar adrenoleucodistrofia cerebral.
La insuficiencia suprarrenal (o incluso una crisis Addisoniana que suponga un riesgo vital) puede ser el síntoma de presentación de la adrenoleucodistrofia en niños y hombres, años o incluso décadas antes a la aparición de los síntomas neurológicos. Un estudio sobre los niños neurológicamente pre-sintomáticos con adrenoleucodistrofia mostró que el 80% de estos niños ya tenían una alteración de la función suprarrenal en el momento del diagnóstico de la adrenoleucodistrofia. Los signos más comunes de la insuficiencia suprarrenal son crónicos o de larga duración, incluyen fatiga, debilidad muscular, pérdida del apetito, pérdida de peso, dolor abdominal y vómito inexplicable. Otros síntomas pueden incluir náuseas, diarrea, baja presión sanguínea (que se ve incluso más reducida cuando la persona se pone en pie, causando mareo o desmayo), irritabilidad y depresión, antojo de comidas con elevado contenido en sal, niveles bajos de azúcar en sangre, dolor de cabeza, o sudoración. Los individuos afectados pueden tener o no un nivel elevado de pigmentación de piel como resultado de la secreción excesiva de la hormona adrenocorticotropa (ACTH).
Mielopatía: Prácticamente todos los pacientes varones con adrenoleucodistrofia que alcanzan la edad adulta desarrollan una mielopatía, generalmente entre los 20-40 años de edad. Los síntomas se limitan a la médula espinal y los nervios periféricos. Inicialmente, la discapacidad neurológica progresa lentamente. El diagnóstico de la adrenoleucodistrofia se realiza raramente durante los primeros 3-5 años de síntomas clínicos, a no ser que otros casos de adrenoleucodistrofia hayan sido localizados con anterioridad en la misma familia. Los pacientes desarrollan una lenta y progresiva disfunción motora debido a la rigidez y debilidad de las piernas. Los individuos también pueden desarrollar disfunción urinaria con micción urgente, que puede progresar hasta incontinencia. Todos los síntomas son progresivos durante años o décadas. La mayoría de pacientes necesitan asistencia para caminar hacia la quinta o sexta década de su vida.
Adrenomieloneuropatía (AMN): La palabra adrenomieloneuropatía se refiere a los pacientes varones tanto con la función suprarrenal alterada como con mielopatía.
Adrenoleucodistrofia cerebral: Los niños y hombres con adrenoleucodistrofia se encuentran en riesgo de desarrollar lesiones desmielinizantes en la sustancia blanca cerebral (adrenoleucodistrofia cerebral). La aparición de adrenoleucodistrofia cerebral nunca ha sido diagnosticada antes de los 3 años de edad. En el pasado, la adrenoleucodistrofia cerebral se consideraba poco frecuente en la adolescencia (4-7%) y la edad adulta (2-5%). Sin embargo, ahora que realizamos el seguimiento de un gran grupo de varones con adrenoleucodistrofia con escáneres de RM anuales, parece ser que estos porcentajes son en realidad más elevados. Actualmente, no podemos predecir si o cuando un paciente desarrollará adrenoleucodistrofia cerebral. Un posible desencadenante ambiental seria el padecer un traumatismo craneal, pero es probable que se requieran otros factores genéticos y ambientales aún desconocidos para el desarrollo de la adrenoleucodistrofia cerebral. Los síntomas de la adrenoleucodistrofia cerebral son, en general, rápidamente progresivos. Un paciente varón recién nacido tiene un riesgo del 35-40% de desarrollar adrenoleucodistrofia cerebral entre las edades de 3 y 18 años. Por lo general, los niños afectados (en edad de asistir a la escuela primaria) inicialmente tienen problemas de comportamiento o déficits de aprendizaje, que se manifiestan como un declive en el rendimiento escolar. Estos primeros síntomas clínicos, son frecuentemente diagnosticados como trastorno por déficit de atención o hiperactividad, lo que puede retrasar el diagnóstico de la adrenoleucodistrofia. En pacientes adultos, los primeros síntomas son frecuentemente psiquiátricos también y pueden asemejarse a depresión o psicosis. En estos pacientes, el diagnóstico de la adrenoleucodistrofia es demorado frecuentemente; especialmente cuando la adrenoleucodistrofia no se encuentra presente en la historia familiar y cuando los síntomas clínicos de insuficiencia adrenal se hallan ausentes. A medida que la enfermedad avanza, los déficits neurológicos se hacen más evidentes. Estos incluyen discapacidad auditiva, disminución de la agudeza visual, debilidad en las extremidades, problemas con la coordinación y convulsiones. Esta etapa es extremadamente rápida y devastadora. Los pacientes afectados pueden perder la capacidad de comprender el lenguaje y caminar en pocos meses. Con el tiempo, los pacientes están postrados en cama, ciegos, incapaces de hablar o responder, lo que requiere cuidados de enfermería a tiempo completo y alimentación por sonda nasogástrica o gastrostomía. Por lo general, la muerte se produce de 2 a 4 años después de la aparición de los síntomas iniciales, o – si el paciente recibe unos buenos cuidados – los pacientes pueden permanecer en este estado vegetativo aparente durante varios años.
Las mujeres con adrenoleucodistrofia: Como en muchas enfermedades ligadas al cromosoma X, se asumió originalmente que las mujeres portadoras del gen afectado para la adrenoleucodistrofia permanecen asintomáticas. Sin embargo, actualmente se ha podido establecer que esta premisa es incorrecta. De hecho, en un estudio reciente se ha demostrado que más de 80% de las mujeres con adrenoleucodistrofia desarrollan síntomas después de la edad de 60 años (para más información consulte la entrada: mujeres con adrenoleucodistrofia). El texto completo de este estudio se puede consultar y descargar (en formato pdf). En general, la aparición de los síntomas neurológicos se produce a una edad más tardía que en los varones con mielopatía; típicamente entre los 40 y los 50 años de edad. La progresión de la enfermedad es generalmente más lenta que en varones. A diferencia del caso de los varones, la incontinencia fecal es una queja frecuente en mujeres con adrenoleucodistrofia. Es importante señalar que la adrenomieloneuropatía en mujeres con adrenoleucodistrofia se diagnostica a menudo como esclerosis múltiple. Tanto el fallo suprarrenal como la adrenoleucodistrofia cerebral son muy raros, menos de 1%, respectivamente. Para más detalles consulte (Females with ALD).
Pruebas
La adrenoleucodistrofia se diagnostica mediante un simple análisis de sangre, que mide los niveles de ácidos grasos de cadena muy larga. Esta prueba es precisa en los hombres, y es ampliamente aceptada como una herramienta altamente precisa para el diagnóstico de varones de todas las edades. Sin embargo, en aproximadamente el 20% de las mujeres con adrenoleucodistrofia el análisis de VLCFA muestra niveles normales y por lo tanto supone un resultado “falso negativo” para el individuo. Una forma de identificar de forma precisa a los pacientes “falsos negativos” es mediante una prueba de ADN. Esta prueba permite la identificación precisa de mujeres con adrenoleucodistrofia mediante pruebas genéticas, y ante resultados normales puede asegurar que una mujer no es un portadora del gen defectuoso para la adrenoleucodistrofia.
Cribado Neonatal
El diagnóstico temprano de la adrenoleucodistrofia es la clave para salvar vidas, debido a que el cribado neonatal permite realizar un monitoreo prospectivo de la función suprarrenal y de la aparición de adrenoleucodistrofia cerebral. Una prueba para el cribado neonatal ha sido desarrollada. Esta prueba detecta niveles elevados de VLCFA (como la C26:0-lysoPC) en gotas de sangre. El 30 de diciembre de 2013, el Estado de Nueva York empezó a realizar las pruebas de cribado para la adrenoleucodistrofia en recién nacidos. En febrero de 2016, la adrenoleucodistrofia fue añadida al Panel de detección uniforme federal recomendado (RUSP) de Estados Unidos. Desde entonces, otros estados y países han empezado a implementar la enfermedad en sus programas de cribado neonatal, o han iniciado los procesos correspondientes destinados a su implementación en sus actuales programas de cribado. Información actualizada y detallada sobre el cribado neonatal de la adrenoleucodistrofia puede ser consultado en la página “Cribado neonatal”.
Investigación
Una amplia investigación sobre la adrenoleucodistrofia se está haciendo en todo el mundo. En 1993, el gen para la adrenoleucodistrofia se identificó a través de los esfuerzos combinados de los doctores Patrick Aubourg y Jean-Louis Mandel en Francia y el Dr. Hugo Moser en los EE.UU., lo que ha abierto nuevas puertas para su posterior estudio. Las actividades de investigación se centran en muchos aspectos, para responder a preguntas fundamentales, como: “¿Cómo influye la acumulación de ácidos grasos de cadena muy larga en la pérdida de la mielina?”; “¿Por qué un paciente desarrolla adrenoleucodistrofia cerebral mientras que otro (que puede incluso ser el hermano del paciente) desarrolla una mielopatía a una edad más avanzada?”
Tratamiento
A día de hoy no existe un tratamiento curativo para la adrenoleucodistrofia.
Terapia sustitutiva de esteroides suprarrenales: La mayoría de pacientes varones desarrollan insuficiencia suprarrenal. La insuficiencia suprarrenal a menudo es la primera manifestación de la adrenoleucodistrofia: Un estudio reveló que el 80% de los niños neurológicamente pre-sintomáticos con adrenoleucodistrofia, que fueron identificados mediante un análisis extendido a la familia, ya presentaban alteraciones de la función suprarrenal a la edad de 4 años. Para estos pacientes, la terapia sustitutiva de esteroides suprarrenales es obligatoria, y puede salvar vidas, pero no tiene ningún efecto sobre los síntomas neurológicos.
Para la mielopatía, que afecta al 85% de todos los pacientes con adrenoleucodistrofia (hombres y mujeres combinados), ninguna terapia curativa está disponible.
Restricción dietética: Debido a que los VLCFA son tóxicos para la mielina, las glándulas suprarrenales y los testículos, se hicieron varios intentos para disminuir las concentraciones plasmáticas de estos. La restricción dietética de la ingesta de VLCFA por sí sola no tiene ningún efecto sobre los niveles plasmáticos de estos.
El aceite de Lorenzo: Los VLCFA se sintetizan principalmente a través de la elongación de la cadena de ácidos grasos más cortos. En el laboratorio, la adición de ácidos grasos monoinsaturados al medio de cultivo de los fibroblastos con mutaciones para la adrenoleucodistrofia, reduce las concentraciones de VLCFA a niveles fisiológicos. Esto puede explicarse debido a que las enzimas implicadas en la síntesis de VLCFA son las mismas encargadas de sintetizar tanto ácidos grasos monoinsaturados como ácidos grasos saturados. Sin embargo, su afinidad por los ácidos grasos monoinsaturados es mayor. Este hallazgo formó la base para una terapia dietética. La administración oral de ácido oleico en forma de triglicéridos (GTO), y ácido erúcico en forma de triglicéridos (GTE) normalizó los niveles de VLCFA en plasma pasado un mes en la mayoría de los pacientes con adrenoleucodistrofia. La combinación de GTO y GTE en una proporción de 4:1 llegó a ser conocido como “aceite de Lorenzo”, en homenaje a Lorenzo Odone, el primer paciente tratado con la mezcla. En aquel entonces, se pensó en el aceite de Lorenzo como una terapia prometedora. Sin embargo, varios ensayos clínicos han demostrado que el aceite no conseguiría mejorar la función neurológica o endocrina ni podría detener la progresión de la enfermedad. Para más detalles consulte (Lorenzo’s oil).
La lovastatina: Se demostró que tiene un efecto sobre los VLCFA. Este hallazgo, sin embargo, no pudo ser reproducido por otros. De hecho, experimentos posteriores mostraron que las estatinas no tenían ningún efecto sobre los niveles de VLCFA del cerebro ni de las glándulas suprarrenales en ratones con adrenoleucodistrofia, e incluso producían una acumulación de VLCFA en estos tejidos. Debido a estos resultados contradictorios, un ensayo clínico controlado con placebo, aleatorizado y a doble ciego se llevó a cabo en el Centro Médico Académico de Ámsterdam para probar el efecto de la lovastatina como una terapia para la reducción de VLCFA en la adrenoleucodistrofia. Los resultados y conclusiones, que fueron publicados en el New England Journal of Medicine, demuestran que el tratamiento con lovastatina produce una pequeña disminución en plasmática de VLCFA, pero no tiene ningún efecto sobre los niveles de estos a nivel celular, ya que los niveles de C26:0 en los eritrocitos y leucocitos no experimentaron cambios. Para más detalles consulte la página (Lovastatin).
Bezafibrato: En la búsqueda de compuestos que podrían reducir los niveles de VLCFA, el bezafibrato, un fármaco utilizado para el tratamiento de la hiperlipidemia, fue identificado como un agente reductor de los niveles de VLCFA. Los experimentos en fibroblastos mostraron que el bezafibrato reducía los niveles de VLCFA inhibiendo directamente la actividad de la elongasa ELOVL1 específica de VLCFA. Un estudio piloto abierto se realizó para evaluar el efecto del bezafibrato en la acumulación de VLCFA en las células sanguíneas de pacientes con adrenoleucodistrofia. Desafortunadamente, el bezafibrato no logró disminuir los niveles de estos ácidos grasos en las células sanguíneas de pacientes con adrenoleucodistrofia. Lo más probable es que esto se debiera a su incapacidad para alcanzar niveles adecuados de fármaco en los pacientes.
Trasplante de médula ósea: En niños y adolescentes en las primeras etapas de la adrenoleucodistrofia cerebral, el trasplante alogénico de células madre hematopoyéticas (TCMH) puede detener la progresión de la desmielinización cerebral en la adrenoleucodistrofia siempre y cuando el procedimiento se realice en una etapa muy temprana de la enfermedad. La eficacia del trasplante se basa en la renovación de las células de la microglía del cerebro deficientes para ALDP por células normales de la microglía que se originan a partir de las células madre de médula ósea del donante. Para más detalles consulte la página (Hematopoietic stem cell transplantation).
Terapia génica: Se prevé que en un futuro no muy lejano, el trasplante de células madre hematopoyéti-cas autólogas (las propias células de la médula ósea del paciente) que habrán sido ge-néticamente corregidas ex vivo (fuera del cuerpo del paciente) con un vector lentiviral antes de reinfundirlas al paciente, podría llegar a ser una opción terapéutica adicional. Este optimismo se basa en los resultados muy alentadores reportados en el año 2009 en los dos primeros pacientes con adrenoleucodistrofia tratados en ensayos clínicos (Cartier et al. 2009) y en los datos recientes del estudio Starbeam, que fueron publicados en octubre de 2017. Más detalles en la página (Gene Therapy for ALD).
Un resumen de 10 minutos de la adrenoleucodistrofia
Producido por Youreka Science en colaboración con ALD Connect, Inc.
Por favor consulta la página web de ALD Connect Educational Videos & Webinars page para más vídeos.
Cribado neonatal
Introducción
Los bebés nacidos con adrenoleucodistrofia son neurológicamente normales en el momento del nacimiento. Sin embargo, el diagnóstico precoz de estos niños puede llevar a la aplicación de intervenciones capaces de salvar sus vidas. Estas incluyen la puesta en marcha de una terapia suprarrenal sustitutiva basada en el uso de esteroides tras la detección de insuficiencia suprarrenal, y la administración de un trasplante alogénico de células madre hematopoyéticas (TCMH) como medio para tratar la adrenoleucodistrofia cerebral. El TCMH puede detener la progresión, que a menudo suele ser fatal, de la desmielinización cerebral, debido a que el procedimiento se realiza en un estadio muy temprano de la enfermedad. Desafortunadamente, esto tan solo puede llegar a ser efectivo durante una estrecha ventana terapéutica, que con frecuencia es pasada por alto. El cribado neonatal da acceso a esta “ventana de oportunidad” y permite iniciar a tiempo estas terapias establecidas.
En febrero de 2016, la adrenoleucodistrofia fue añadida al Panel de detección uniforme federal recomendado (RUSP) en EE.UU., que corresponde a la lista federal de todas las enfermedades genéticas recomendadas para los programas de cribado estatales. El estado de Nueva York inició el cribado de la adrenoleucodistrofia en recién nacidos el 30 de diciembre de 2013. Desde entonces, otros estados empezaron a incluir la adrenoleucodistrofia dentro de sus programas de cribado neonatal (Fig 1). En EE.UU., muchos más estados cuentan con la aprobación legislativa. Fuera de EE.UU., el Ministerio de Sanidad en los Países Bajos, ha aprobado la inclusión de la adrenoleucodistrofia al programa de cribado neonatal y un programa piloto dará su inicio en el año 2019. Se prevé que el cribado neonatal de la adrenoleucodistrofia empezará a aplicarse en estos estados y países tan pronto como se disponga de los correspondientes recursos presupuestarios, procedimientos de prueba y protocolos de seguimiento.

Figura 1: El mapa muestra aquellos estados de EE.UU. que ya han iniciado un programa de cribado para la adrenoleucodistrofia.
Criterios para la inclusión en el programa de cribado
Existe un amplio consenso internacional en cuanto a los criterios que debe cumplir una enfermedad para poder ser incluida dentro de un programa de cribado neonatal.
- El diagnóstico precoz de la enfermedad debe ser directamente ventajoso para el recién nacido. Deben existir beneficios sustanciales para la salud, derivados de una intervención temprana en enfermedades graves que consten de un curso natural conocido.
- La prueba de cribado debe ser de buena calidad. El correspondiente ensayo debe presentar una especificidad y sensibilidad elevadas, lo que se traduce en tasas reducidas tanto de falsos positivos como de falsos negativos.
 Historia
Historia
En el año 2004, en la reunión del comité asesor nacional para la evaluación de recién nacidos, el Dr. Hugo Moser sugirió añadir la adrenoleucodistrofia al RUSP de los Estados Unidos. El único problema fue que, en aquel momento, no se encontraba disponible ninguna prueba válida para el cribado neonatal de la enfermedad. Para poder salvar esta dificultad, recaudó fondos y contrató a un equipo de investigadores en el instituto Kennedy Krieger (Baltimore, MD) con el objetivo de identificar un biomarcador adecuado y desarrollar una prueba utilizando la espectrometría de masas en tándem (MS/MS). En 2006, el equipo informó de la identificación de la C26:0-lisofosfatidilcolina (C26:0-LPC) en muestras secas de sangre venosa postnatal (Dried Blood Spots, DBS) de varones con adrenoleucodistrofia (Hubbard et al. 2006). Durante los años siguientes, los científicos continuaron trabajando en la mejora de los análisis (Hubbard et al. 2009; Theda et al. 2014). Junto con los investigadores en la Clínica Mayo (Rochester, Minnesota), se desarrolló un método de alto rendimiento para el análisis de la C26:0-LPC (Haynes and De Jesús 2012; Turgeon et al. 2015). En 2013, este método fue validado utilizando 100.000 muestras secas de sangre anónimas.

La ley de Aidan (Aidan’s Law)
En Abril de 2012, tras la muerte de su hijo, Aidan, el cuál padecía adrenoleucodistrofia cerebral pero fue diagnosticado demasiado tarde, la familia Seeger redactó y dio apoyo a la aprobación de la ley de Aidan en el estado de Nueva York. El proyecto de ley fue aprobado en Febrero del año 2013 y se convirtió en ley en Marzo de 2013. El 30 de Diciembre de 2013, el laboratorio de cribado neonatal del estado de Nueva York empezó a evaluar recién nacidos para la detección de la adrenoleucodistrofia.
El estado de Nueva York
Durante sus primeros tres años, el estado de Nueva York ha evaluado a más de 700.000 recién nacidos y ha identificado a 45 bebés con adrenoleucodistrofia: 22 niños y 23 niñas. Basándonos en estas cifras, la incidencia de la adrenoleucodistrofia en el nacimiento es de 1 por cada 15.000 recién nacidos examinados. Cuando se identifica a un recién nacido con adrenoleucodistrofia, se notifica al médico de cabecera de la familia y esta se deriva a un genetista clínico para realizar la confirmación del diagnóstico, junto con un asesoramiento genético para ofrecer servicios de apoyo y detección de otros miembros de la familia con riesgo de padecer adrenoleucodistrofia (detección extendida a la familia).
Para los varones, es esencial iniciar una monitorización seriada mediante una resonancia magnética cerebral para detectar de forma temprana las primeras evidencias de la aparición de la adrenoleucodistrofia cerebral; y para iniciar las pruebas de función suprarrenal con el objetivo de detectar la insuficiencia suprarrenal. La evaluación integral a nivel neurológico, neuropsicológico, neuroradiológico, y a nivel de la función suprarenal es necesaria debido a que no existe ninguna prueba que prediga el desarrollo clínico de un bebé nacido con una mutación característica de la adrenoleucodistrofia.
La prueba de cribado neonatal
La prueba de la C26:0-LPC y sus reactivos asociados puede diferir ligeramente entre laboratorios. No obstante, en todos los casos, el diagnóstico de la adrenoleucodistrofia es llevado a cabo usando un algoritmo de 3 niveles (Fig 2). El primer nivel consiste en un análisis estándar de alto rendimiento mediante MS/MS de la C26:0-LPC. Las muestras que contienen una concentración elevada de C26:0-LPC son analizadas posteriormente en el segundo nivel, utilizando HPLC-MS/MS. Esta prueba es más específica, pero también requiere algo más de tiempo. Aquellas muestras que aún muestren concentraciones elevadas de C26:0-LPC, serán analizadas en el tercer nivel, en el cuál se realizará la secuenciación del gen ABCD1.
 Figura 2: Los principios de la detección en 3 niveles para la adrenoleucodistrofia.
Figura 2: Los principios de la detección en 3 niveles para la adrenoleucodistrofia.
Desafíos
En diferentes países existen desafíos significativos y debates éticos continuos en cuanto a la implementación del cribado neonatal de la adrenoleucodistrofia.
- El primer criterio para la inclusión en un programa de cribado neonatal, que establece que el diagnóstico precoz debe ser directamente ventajoso para el recién nacido, puede ser causa de preocupaciones éticas. En la adrenoleucodistrofia, aproximadamente uno de cada tres niños varones desarrollará adrenoleucodistrofia cerebral entre los 3 y los 18 años. Sin embargo, los dos tercios restantes de varones con adrenoleucodistrofia desarrollaran adrenomieloneuropatía (AMN) durante la edad adulta, la cuál se caracteriza por espasticidad de las extremidades, disfunción motora e incontinencia. El AMN se trata de forma sintomática. La ausencia de marcadores en el laboratorio o de otras herramientas biológicas dificulta la predicción del desarrollo de la enfermedad para aquellos individuos afectos, hecho que puede incrementar el riesgo en cuanto a la aplicación de intervenciones médicas innecesarias.
- El cribado neonatal también permite identificar a aquellas niñas portadoras de un gen de la adrenoleucodistrofia alterado. Las mujeres con adrenoleucodistrofia tienen una probabilidad <<1% de desarrollar insuficiencia suprarrenal o adrenoleucodistrofia cerebral, y consecuentemente no existe ningún beneficio directo para la salud para una niña recién nacida con adrenoleucodistrofia, ya que no podría ser tratada mediante TCMH o terapia hormonal suprarrenal. Aproximadamente, un 80% de las mujeres con adrenoleucodistrofia acabaran desarrollando una mielopatía a la edad de 60 años.
- En varios países existe un debate creciente entre la comunidad científica y entre las organizaciones de pacientes con respecto a la inclusión de ciertas enfermedades no tratables en los programas de cribado neonatal. Como se menciona anteriormente, un diagnóstico precoz, en última instancia, debe ser capaz de proporcionar un beneficio directo para la salud del recién nacido. Esto puede no ser evidente en el caso de una enfermedad que pueda ser diagnosticada pero no tratada.
- En ocasiones el cribado neonatal puede identificar enfermedades que se encuentran más allá del objetivo de la prueba. La prueba de cribado neonatal de C26:0-LPC también permite identificar enfermedades no tratables asociadas con un incremento en los niveles de C26:0-LPC (Fig 2). Entre estas se incluyen: el espectro del síndrome Zellweger; los trastornos en la oxidación peroxisomal de los ácidos grasos, causados por un defecto en la acyl-CoA oxidasa 1 (ACOX1) peroxisomal o bien en la proteína multifuncional (HSD17B4); el “Síndrome de deleción contigua ABCD1 DXS1357E” (CADDS); la deficiencia del dominio de unión a acyl-CoA que contiene la proteína 5 (ACBD5); y el síndrome de Aicardi Goutières.
- En algunos escenarios, otras ventajas, más allá de aquellas que se encuentran destinadas a mejorar la salud del recién nacido pueden ser consideradas. Algunas de estas pueden beneficiar al recién nacido, como por ejemplo un proceso diagnóstico más rápido. No obstante, la mayoría de ventajas suponen un claro beneficio para la familia, como la posibilidad de realizar una detección extendida para poder identificar a otros miembros de la familia en riesgo; y el ajuste de la vida familiar para poder hacer frente a las consecuencias de la enfermedad. Además, los progenitores podrían beneficiarse también del cribado enfocado a una enfermedad para la cual no existe un tratamiento efectivo ya que este conocimiento confiere a los padres información que podrían aplicar de cara a la toma de futuras decisions reproductivas. Sin embargo, existen también claras desventajas. El diagnóstico de una enfermedad no tratable podría proyectar una sombra o stigma sobre los primeros años de vida y la infancia del recién nacido.
Como resultado de ser añadido al RUSP, se prevé que el cribado neonatal de la adrenoleucodistrofia sea iniciado en un número creciente de estados en los EE.UU. en los próximos años, así como en otros países. Por ejemplo, los Países Bajos empezaran un estudio piloto para el cribado neonatal de la adrenoleucodistrofia en el 2019. Estas medidas mejoraran de forma significativa el desarrollo clínico de cientos de recién nacidos con adrenoleucodistrofia, sus parientes biológicos y sus seres queridos.
Guía para el asesoramiento
Authors: Marc Engelen, M.D., Ph.D. and Stephan Kemp, Ph.D.
(Traducido por Nerea Montedeoca Vázquez (Biomedicine student))
Introducción
El espectro clínico en varones con adrenoleucodistrofia varía desde una insuficiencia suprarrenal aislada y una lenta y progresiva mielopatía hasta una severa desmielinización cerebral (adrenoleucodistrofia cerebral). La mayoría de las mujeres afectadas desarrollaran síntomas hacia la edad de 60 años. En ausencia de una correlación genotipo-fenotipo, no es posible predecir el curso de la enfermedad; ni siquiera dentro de una misma familia. Este artículo se centra en el manejo de los pacientes con adrenoleucodistrofia y constituye una guía para los clínicos que se encuentren con pacientes afectos de esta enfermedad enormemente compleja.
El diagrama de flujo inferior resume las recomendaciones para el seguimiento de niños y varones adultos con adrenoleucodistrofia.
>
<h3Niños con adrenoleucodistrofia
El seguimiento de niños con adrenoleucodistrofia es importante por dos razones: 1) la detección precoz de la insuficiencia suprarrenal y 2) la detección temprana de la adrenoleucodistrofia cerebral para proponer un trasplante alogénico de células madre hematopoyéticas (TCMH) si un donante HLA-compatible o un cordón umbilical se hallan disponibles. A pesar de la existencia de un riesgo de mortalidad significativo, el TCMH alogénico constituye hasta el momento, la única intervención terapéutica capaz de detener la progresión de la desmielinización cerebral en la adrenoleucodistrofia, siempre y cuando el procedimiento se realice de forma muy temprana, es decir, cuando los niños afectados tengan leves o ningún síntoma a causa de la enfermedad cerebral desmielinizante.
En el futuro, trasplántelos trasplantes autólogos de células madre hematopoyéticas que hayan sido genéticamente corregidas mediante un vector lentiviral antes de su reinfusión, podrían convertirse en una alternativa al TCMH alogénico, una vez que los muy alentadores resultados obtenidos en los dos primeros pacientes tratados se hayan extendido a un número mucho mayor de pacientes con adrenoleucodistrofia cerebral.
Si estos niños no padecen la enfermedad de Addison, es recomendable que un endocrino los evalúe anualmente para insuficiencia suprarrenal, a través de la medición de los niveles de ACTH en plasma y aplicando una prueba para la hormona ACTH. La terapia sustitutiva basada en esteroides podría entonces ser iniciada en caso de ser necesario.
Los niños sin déficits neurológicos deberían ser estrechamente monitoreados en busca de signos radiológicos de adrenoleucodistrofia cerebral. La adrenoleucodistrofia cerebral acaecido en la infancia (CCALD) no ha sido detectado antes de la edad de 2,5 años. Recomendamos una resonancia magnética (RM) del cerebro cada 6 meses en niños con edades comprendidas entre los 3 y los 12 años con el fin de detectar signos tempranos de CCALD. Si aparecen síntomas que sugieran adrenoleucodistrofia cerebral (como por ejemplo una disminución del rendimiento escolar) la RM debería ser realizada a la primera oportunidad que se presente, pero según nuestra propia experiencia, la detección de anomalías cerebrales en una RM precede a cualquier disfunción cognitiva detectable desde como mínimo 6 meses hasta 1 año. Más allá de los 12 años de edad, la incidencia de CCALD disminuye. aun así, una RM deberá ser realizada anualmente o incluso antes si aparecen nuevos síntomas.
Es importante detectar la adrenoleucodistrofia cerebral tan pronto como sea posible, preferentemente en su fase asintomática con tan solo anomalías radiológicas moderadas para poder debatir sobre la posibilidad de realizar un TCMH alogénico. En consecuencia, si una RM cerebral mostrase anomalías, incluso siendo estas muy limitadas como por ejemplo un incremento en la intensidad de señal en las secuencias T2 o FLAIR en el esplenio o en la rodilla del cuerpo calloso, debería repetirse la RM cerebral en un período máximo de 3 meses para poder evaluar la progresión de la enfermedad y, en particular, para poder identificar la potenciación del gadolinio en los bordes de las zonas lesionadas. Debido a que la enfermedad puede presentar una progresión muy rápida, se recomienda encarecidamente el hecho de contemplar y debatir la posibilidad de realizar un TCMH alogénico tan pronto como las anomalías típicas de la adrenoleucodistrofia cerebral sean detectadas. Tras un trasplante exitoso, las lesiones en la RM pueden estabilizarse e incluso experimentar una regresión. Los resultados del tratamiento son mejores cuanto más pronto se inicia este.
Varones adultos con adrenoleucodistrofia
El seguimiento de los varones con adrenoleucodistrofia es importante de cara a una detección temprana de insuficiencia suprarrenal. Si estos no padecen la enfermedad de Addison se recomienda que un endocrino los evalúe anualmente para la disfunción adrenocortical mediante la medición de los niveles de ACTH en plasma y la realización de una prueba para la hormona ACTH. La terapia sustitutiva basada en esteroides podría entonces ser iniciada en caso de ser necesario.
Para aquellos varones adultos con o sin signos de mielopatía, recomendamos una evaluación anual o bianual realizada por un neurólogo con el objetivo de detectar síntomas de mielopatía y de administrar tratamiento sintomático si fuera necesario (por ejemplo, medicación para la espasticidad). La derivación a un médico de rehabilitación y a un urólogo a menudo será necesaria.
Los varones adultos pueden llegar a desarrollar adrenoleucodistrofia. En nuestros centros, ofrecemos una RM cerebral de forma anual. Actualmente, no existe ningún tratamiento probado para tratar la adrenoleucodistrofia cerebral en adultos. Parece probable que el TCMH alogénico sea también efectivo en adultos con un estadio temprano de adrenoleucodistrofia cerebral, pero no existen estudios o casos publicados describiendo este tratamiento. En general, tendemos a considerar un TCMH alogénico en un paciente adulto con un estadio temprano de adrenoleucodistrofia cerebral, tras asesorar cuidadosamente al paciente sobre la falta de evidencia para el tratamiento y el riesgo del procedimiento, el cual es significativamente mayor que en el caso de los niños.
Para la afectación de la médula espinal, aún no se encuentra disponible ninguna terapia efectiva que modifique el curso de la enfermedad. Aunque el aceite de Lorenzo parecía muy prometedor, numerosos ensayos abiertos han demostrado que la enfermedad progresa incluso cuando los niveles de ácidos grasos de cadena muy larga (VLCFA) son normalizados por el tratamiento con aceite de Lorenzo. Un gran ensayo clínico aleatorizado y utilizando placebo fue diseñado con el fin de proporcionar una respuesta definitiva a esta cuestión. Desafortunadamente, tuvo que ser abortado por el comité de control de seguridad antes de ser completado, debido a los supuestos efectos secundarios del tratamiento con placebo. También existe un estudio retrospectivo que sugiere que si niños presintomáticos inician un tratamiento con aceite de Lorenzo, este podría retrasar la aparición de síntomas neurológicos. Consideramos que la evidencia científica apoyando la eficacia del aceite de Lorenzo es débil, y por esto no ofrecemos este tratamiento a nuestros pacientes. Aun así, un seguimiento habitual de los pacientes con mielopatía continúa siendo importante, principalmente para poder proporcionar un tratamiento sintomático.
Mujeres con ALD
Las mujeres con ALD deberían ser evaluadas para el desarrollo de síntomas neurológicos. Dado que las mujeres con ALD muy raramente desarrollan insuficiencia suprarrenal o alguna implicación cerebral, la evaluación periódica de la función suprarrenal y la realización de RM cerebrales no son necesarias. Una mayor concienciación entre los médicos de que las mujeres pueden desarrollar síntomas neurológicos es importante de cara al asesoramiento, pero también para evitar pruebas diagnósticas innecesarias y diagnósticos erróneos. Sabemos de casos de mujeres con ALD quienes pasaron por una laminectomía cervical por una supuesta mielopatía cervical espondilogénica. Para aquellas mujeres síntomáticas con adrenoleucodistrofia, recomendamos (así como para aquellos varones adultos con ALD) una evaluación anual realizada por un neurólogo para debatir la indicación de la rehabilitación, la derivación a un urólogo y el tratamiento de la espasticidad y el dolor neuropático.
El texto en esta página es un resumen del artículo científico: “X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management”. (“Adrenoleucodistrofia ligada al X (X-ALD): Presentación clínica y pautas para el diagnóstico, seguimiento y gestión”), publicado en acceso abierto en el “Orphanet Journal of Rare Diseases (para facilitar la lectura, las referencias han sido eliminadas). El texto completo en inglés puede ser accedido y descargado (como pdf).
Diagnóstico de ALD
Authors: Hugo W Moser, M.D., Ann B Moser, B.A., Steven J Steinberg, Ph.D. and Stephan Kemp Ph.D.
(Traducido por Nerea Montedeoca Vázquez (Biomedicine student))
Diagnóstico clínico
El diagnóstico de la adrenoleucodistrofia se debe considerar en cuatro situaciones clínicas distintas:
- Niños con síntomas de trastorno por déficit de atención, que además muestran signos de demencia, trastorno progresivo de la conducta, pérdida de la visión, dificultad para comprender el lenguaje hablado, deterioro de la escritura, falta de coordinación, u otros trastornos neurológicos.
- Jóvenes o varones de mediana edad con trastornos motores progresivos, rigidez en las piernas o debilidad, anomalías en el control de los esfínteres y disfunción sexual, con o sin insuficiencia suprarrenal o déficits cognitivos o de comportamiento.
- Todos los varones con insuficiencia adrenocortical (suprarrenal) primaria, con o sin evidencias de anomalías neurológicas.
- Mujeres de mediana edad o mayores con paraparesia progresiva, anomalías en el control de los esfínteres y trastornos sensoriales que afecten principalmente a las piernas. Puede ser difícil establecer el diagnóstico de adrenoleucodistrofia en una mujer con una historia familiar negativa. El diagnóstico se basa en las características clínicas, frecuentemente paraparesia espástica progresiva, y un panel de pruebas de laboratorio.
Neuroimagen
La resonancia magnética cerebral (RM) es siempre anormal en varones neurológicamente sintomáticos y, a menudo proporciona la primera ventaja de diagnóstico. En aproximadamente el 85% de los individuos afectados, la RM muestra un patrón característico de la señal simétrica potenciada en T2 en la región parieto-occipital con un incremento del contraste en el margen externo.

Ácidos grasos de cadena muy larga
Hombres: La prueba de laboratorio más importante es la medición de la concentración de ácidos grasos de cadena muy larga (VLCFA) en plasma. Los niveles de VLCFA se encuentran elevados en más del 99,9% de los hombres con adrenoleucodistrofia de todas las edades, independientemente de la presencia o ausencia de síntomas clínicos. Los tres parámetros analizados son: la concentración de C26:0, la relación de C24:0/C22:0, y la relación de C26:0/C22:0. La Tabla 1 muestra los resultados medios para los controles normales, varones afectados y mujeres [Valianpour et al., 2003].
El Análisis de VLCFA es extremadamente especializado y, por lo tanto, se lleva a cabo sólo en unos pocos laboratorios en todo el mundo.
| Controles Normales | Hombres con ALD | Mujeres con ALD | |
| C26:0 µmol/L | 0.67 +/- 0.13 | 2.94 +/- 0.87 | 1.54 +/- 0.72 |
| Relación C24:0/C22:0 | 0.86 +/- 0.13 | 1.52 +/- 0.21 | 1.18 +/- 0.15 |
| Relación C26:0/C22:0 | 0.01 +/- 0.003 | 0.05 +/- 0.02 | 0.02 +/- 0.01 |
| Tabla 1: Concentraciones de VLCFA determinadas en controles y pacientes con adrenoleucodistrofia mediante espectrometría de masas por ionización por electrospray (ESI-MS) (Valianpour et al. 2003). | |||

Importante comentario de la Dra. Ann Moser: El aceite de Lorenzo, una mezcla de ácidos oleico y erúcico, todavía se utiliza en algunos países para normalizar los niveles de VLCFA. El laboratorio de enfermedades peroxisomales en el instituto Kennedy Krieger en Baltimore estudia de manera rutinaria el ácido erúcico (C22:1) cuando se miden los niveles de VLCFA en plasma. Algunos aceites utilizados en la cocina, como el aceite de semilla de mostaza, tienen, de manera natural, niveles elevados de ácido erúcico y, por lo tanto, este hecho puede dar lugar a una elevación similar a la observada durante el tratamiento con aceite de Lorenzo.
Mujeres con ALD: El aumento de la concentración de VLCFA en plasma y/o cultivos de fibroblastos de la piel está presente en aproximadamente el 85% de las mujeres con ALD. Entre el 15-20% de las mujeres identificadas con adrenoleucodistrofia tienen una concentración plasmática normal de VLCFA. Los resultados medios de los niveles plasmáticos de VLCFA obtenidos de mujeres con adrenoleucodistrofia se muestran en la Tabla 1. La función discriminante empleada en el estudio de (Moser AB et al. 1999) no es capaz de distinguir a todas las mujeres con adrenoleucodistrofia del rango normal de los controles (véase la Figura). Las mujeres deben ser sometidas a pruebas genéticas cuando se sospecha de adrenoleucodistrofia y las concentraciones de VLCFA son normales.
Análisis de mutaciones
El gen ABCD1 es el único gen asociado con la adrenoleucodistrofia. Más de 800 mutaciones diferentes se han identificado en ABCD1 [The ALD Mutation Database]. Muchas familias con adrenoleucodistrofia tienen una mutación única. Todas las mutaciones patogénicas identificadas en el gen ABCD1 se encuentran catalogadas en este sitio web.
Boehm y sus colegas han desarrollado y validado una prueba diagnóstica robusta de ADN para la adrenoleucodistrofia mediante el uso de la amplificación genómica no anidada del gen de la adrenoleucodistrofia, seguida de la secuenciación y análisis con cebadores (primers) fluorescentes. Este método cubre, en 10 amplicones independientes, todos los exones codificantes y las uniones intrón-exón que los flanquean [Boehm et al., 1999]. Este protocolo proporciona un medio altamente eficaz para determinar el estado de heterocigosidad en las mujeres con riesgo de transmitir la adrenoleucodistrofia y es aplicable a un laboratorio de diagnóstico clínico. Este método se ha convertido en el análisis diagnóstico (basado en la secuenciación) de elección para muchos laboratorios en todo el mundo.
Asesoramiento Genético
La adrenoleucodistrofia se hereda como un rasgo ligado al cromosoma X. El gen ABCD1 es el único gen asociado con la adrenoleucodistrofia.
Los padres de un paciente de sexo masculino o femenino: Alrededor del 93% de los casos índice han heredado este defecto genético de uno de los padres; por lo que es probable que otros miembros de la familia, como por ejemplo hermanos y primos biológicos, también se encuentren afectados. Sin embargo, en aproximadamente el 7% de los casos, las personas con adrenoleucodistrofia sufrieron una mutación espontanea, también denominada “de novo”. En estos casos, no habrá otros miembros de la familia con el gen mutado. Es conveniente analizar los niveles plasmáticos de VLCFA en las madres de los hombres y mujeres afectadas y en los padres de las mujeres afectadas (Figura inferior). Cuando la mutación causante de la enfermedad haya sido identificada en un miembro afectado de la familia, el análisis de mutación del gen ABCD1 se puede utilizar para evaluar a los padres y para el cribado extendido a toda la familia.
Hermanos de un paciente índice (probando): El riesgo para los hermanos depende del estatus genético de los padres, que puede ser determinado mediante el análisis genealógico, la medición de los niveles de VLCFA, y pruebas de genética molecular. Si la madre de la persona afectada es portadora, la posibilidad de transmitir la mutación causante de la enfermedad en cada embarazo es del 50%. Los hermanos y hermanas que hereden la mutación se encontraran afectados. Si el padre de la persona afectada presenta una mutación causante de la enfermedad en el gen ABCD1, todas sus hijas se encontraran afectadas y ninguno de los hijos varones se verá afectado. Si ninguno de los padres es portador, el riesgo para los hermanos de un individuo afectado es bajo.
Descendencia de un individuo afectado: Los varones que se encuentren afectados trasmitirán la mutación en ABCD1 a todas sus hijas pero a ninguno de sus hijos.
Las mujeres afectadas tienen un 50% de probabilidades de transmitir la mutación en ABCD1 en cada embarazo. Los hijos varones que hereden la mutación se encontraran afectados; las hijas que hereden la mutación serán portadoras y muy probablemente desarrollaran síntomas en la edad adulta.

Figura 2: (Izquierda) Si una mujer es portadora del gen defectuoso para la adrenoleucodistrofia, tendrá las siguientes probabilidades con cada recién nacido: cuando el neonato es una niña, existe una probabilidad del 50% de que la hija reciba el gen afectado en la adrenoleucodistrofia y un 50% de probabilidades de que la hija no se vea afectada. En caso de que el hijo sea un niño, hay un 50% de probabilidades de que el hijo tenga adrenoleucodistrofia y un 50% de probabilidades de que no se vea afectado. (Derecha) Para un trastorno ligado al cromosoma X, tal como la adrenoleucodistrofia, si un hombre afectado tiene hijos, entonces ninguno de sus hijos varones se verán afectados por la enfermedad (el padre siempre pasa su cromosoma Y a sus hijos). Sin embargo, todas sus hijas heredaran el gen defectuoso de la adrenoleucodistrofia (el padre siempre pasa su único cromosoma X (afectado) a sus hijas).
Identificación de los individuos heterocigotos
La realización de las pruebas para identificar portadores en familiares de mujeres en riesgo consta de dos pasos. La determinación de la concentración plasmática de VLCFA se realiza primero; si esta resulta ser anormal, la mujer se encontrará afectada. Debido a que alrededor del 15% de las mujeres con adrenoleucodistrofia tiene una concentración plasmática normal de VLCFA, se deberían utilizar pruebas de genética molecular para evaluar a aquellas mujeres con una concentración normal. Esto es más fácil si la mutación en ABCD1 causante de la enfermedad ya se ha identificado en la familia.
Análisis extendido a la familia
Dependiendo de su sexo, relación familiar, y del estado de portador de los padres del individuo estudiado, las tías y los tíos del individuo afectado y los hijos de estos pueden correr el riesgo de ser portadoras o encontrarse afectados.
La evaluación de los familiares en riesgo es importante para la gestión y el asesoramiento genético, pero se implementa a menudo de manera insuficiente. Hay varios factores que pueden contribuir a una evaluación insuficiente:
- El establecimiento del diagnóstico en un individuo afectado con discapacidad grave puede ser devastador para una familia. Las preocupaciones inmediatas pueden opacar la evaluación oportuna de los miembros de la familia.
- Las pruebas genéticas moleculares se encuentran disponibles para su aplicación en el ámbito clínico desde hace relativamente poco tiempo; pero su disponibilidad puede no ser conocida todavía de forma generalizada.
- Las compañías de seguros podrían no cubrir el coste de las pruebas a aquellos miembros de la familia en situación de riesgo.
- Algunos miembros de la familia en riesgo podrían optar por no ser evaluados, debido al miedo de que un resultado positivo pusiera en peligro su capacidad de obtener o mantener la cobertura del seguro médico.
- Los individuos podrían tener un conocimiento incompleto sobre los miembros de la familia en situación de riesgo y podrían no querer informarles sobre este riesgo.
Análisis prenatal
El diagnóstico prenatal para detectar un posible feto varón afectado puede ser ofrecido a las mujeres cuyo estado de portadora haya sido claramente confirmado por el análisis genético del gen ABCD1. En algunos países, la determinación prenatal no invasiva del sexo del feto utilizando el ADN fetal libre presente en la sangre materna, se puede llevar a cabo a las 7 semanas de embarazo. Esta prueba se basa en la detección de secuencias del cromosoma Y mediante técnicas de PCR. Si el feto es un varón, el análisis mutacional de ABCD1 se puede realizar en una muestra fresca de vellosidades coriónicas (Corionic villus sample, CVS) a las 11-13 semanas de embarazo. Alternativamente, la determinación del sexo fetal se puede realizar por técnicas de citogenética convencional en una muestra de CVS a las 11-13 semanas de embarazo y después de eso, si el feto resultara ser un varón, se podría realizar un análisis mutacional para ABCD1. El análisis de mutaciones tarda unos pocos días. El diagnóstico prenatal también se puede realizar en células del líquido amniótico a las 15-18 semanas de gestación. Este enfoque, sin embargo, requiere al menos de unas 2-3 semanas adicionales de cultivo de células del líquido amniótico con el fin de generar material celular suficiente para el análisis molecular. En caso de que la mutación en ABCD1 aún no haya sido identificada en la familia, pero el estado de portador de la mujer se haya establecido de manera clara bioquímicamente mediante la demostración del aumento de los niveles de VLCFA plasmáticos, el diagnóstico prenatal de un feto varón tendrá que ser realizado a través de la medición de los niveles de VLCFA en las células cultivadas de CVS o en células del líquido amniótico. El diagnóstico genético preimplantacional también se puede ofrecer en algunos países para casos concretos. Por lo general, esto se lleva a cabo cuando una mujer ha tenido al menos dos diagnósticos prenatales que han acabo desembocando en la interrupción del embarazo debido a que el feto era un varón con adrenoleucodistrofia. Un número significativo de mujeres con adrenoleucodistrofia desarrolla una mielopatía en la edad adulta. El diagnóstico prenatal de un feto femenino para establecer si este presenta adrenoleucodistrofia podría ser ofrecido de manera individual. Esta situación se produce con mayor frecuencia cuando el padre o la madre embarazada tiene síntomas clínicos graves.
Cribado neonatal
El diagnóstico temprano de la adrenoleucodistrofia es clave para salvar vidas, debido a que el cribado neonatal permite monitorizar de manera prospectiva la función suprarrenal y la aparición de la adrenoleucodistrofia cerebral. Una prueba para el cribado neonatal ya ha sido desarrollada. Esta, es capaz de detectar niveles elevados de VLCFA (como C26:0-lisoPC) en gotas de sangre. El 30 de diciembre de 2013, el Estado de Nueva York comenzó a realizar el cribado neonatal para la adrenoleucodistrofia. En febrero de 2016, la adrenoleucodistrofia fue añadida al Panel de detección uniforme federal recomendado (RUSP) en Estados Unidos. Desde entonces, otros estados y países han iniciado programas de cribado neonatal o han empezado procesos destinados a añadir la adrenoleucodistrofia a sus propios programas de cribado. Información detallada y actualizada sobre el programa de cribado neonatal de la adrenoleucodistrofia puede ser consultada en la página “Cribado neonatal”.
Mujeres con ALD
Authors: Marc Engelen, M.D., Ph.D., Stephan Kemp, Ph.D., Björn M. van Geel, M.D., Ph.D.
(Traducido por Nerea Montedeoca Vázquez (Biomedicine student))
Introducción
Durante muchos años la adrenoleucodistrofia ha sido considerada como una enfermedad que sólo afecta a niños y hombres. Sin embargo, ha quedado claro que las mujeres portadoras de una mutación en el gen ABCD1 también desarrollan síntomas neurológicos. Sólo en muy raras ocasiones las mujeres con ALD desarrollan una función deficiente de las glándulas suprarrenales o la forma cerebral de la enfermedad. Las mujeres con adrenoleucodistrofia son, por lo tanto, un grupo inequívoco entre los pacientes con esta enfermedad. Esta página se ocupa de los síntomas que las mujeres con adrenoleucodistrofia pueden llegar a desarrollar, y sobre qué se puede hacer al respecto.
Contrariamente a los conceptos anteriores, se ha demostrado claramente que los trastornos ligados al cromosoma X no sólo afectan a los hombres, sino que, las mujeres con los genes mutados, aunque en menor medida, también pueden verse afectadas. La adrenoleucodistrofia no es una excepción. Las mujeres con adrenoleucodistrofia también se consideran como pacientes, aun así, esto es menos conocido. Con frecuencia, los síntomas se atribuyen a otras causas, y por lo tanto, el tratamiento sintomático puede retrasarse. En nuestra experiencia en los Países Bajos, las mujeres aun no diagnosticadas con adrenoleucodistrofia habían sido sometidas a procedimientos quirúrgicos (reemplazo de cadera, operaciones de la columna cervical), algunos más de una vez. Más adelante, se demostró que los síntomas de la enfermedad de la médula espinal eran debidos a la adrenoleucodistrofia. En el peor de los casos, los estados de portador de adrenoleucodistrofia que no fueron reconocidos impidieron un asesoramiento genético, un diagnóstico adecuado y el tratamiento de otros miembros de la familia.
Historia de una portadora
Cuando tenía 38 años de edad, la señora H., nacida en los Países Bajos, se trasladó a los EE.UU., junto con su familia. Se había dado cuenta de que mantener el equilibrio progresivamente se hizo más y más difícil. Además, comenzó a sentir rigidez en sus piernas. Ella tuvo problemas con la función de la vejiga durante varios años, pero asumió que dar a luz a sus dos hijos había causado esto. Uno de sus primos, que en un principio se pensaba que tenía un tumor cerebral, murió a la edad de 13 años. Otro primo, que tenía problemas progresivos al caminar, fue diagnosticado con esclerosis múltiple. Después de leer un artículo sobre la adrenoleucodistrofia en una revista popular en 1985, reconoció sus síntomas y los de sus primos, y decidió ponerse en contacto con el Dr. Hugo Moser en el Instituto Kennedy Krieger en Baltimore, MD. Se le tomó una muestra de sangre, y su análisis reveló una elevación de los ácidos grasos de cadena muy larga (VLCFAs), confirmando así el diagnóstico de la adrenoleucodistrofia.
Cuando la señora H. cumplió 55 años, visitó el Centro Médico Académico de Ámsterdam, Holanda. Al parecer, algunos de sus parientes habían sido reportados en la literatura médica, pero al parecer muchos aún no se habían hecho pruebas para la adrenoleucodistrofia. Algunos optaron por no someterse a las pruebas, pero a muchos de ellos nunca se les había ofrecido el análisis bioquímico para medir los ácidos grasos de cadena muy larga. En el examen físico se detectó un incremento en el tono muscular de las piernas, además de algo de debilidad. La sensación era anormal, el tacto ligero, la vibración y el sentido de la posición estaban deteriorados en piernas y pies. Los reflejos de los tendones fueron enérgicos en las piernas y las respuestas plantares resultaron extensoras.
El estudio de imágenes por resonancia magnética (IRM) del cerebro y la médula espinal eran normales. Asesoramiento genético y análisis de sangre fueron ofrecidos a los familiares.
Retrospectivamente, la adrenoleucodistrofia cerebral infantil y la variante de la adrenoleucodistrofia con afectación de la médula espinal (mielopatía) fueron diagnosticadas en sus primos. Ella fue derivada a un médico de rehabilitación y a un fisioterapeuta.
Junto con su marido, el director de la organización holandesa de pacientes, ella apoyó a las familias y los pacientes recientemente diagnosticados durante muchos años, hasta que falleció de forma inesperada y repentina, debido a una enfermedad no relacionada con la adrenoleucodistrofia.
Los síntomas de las mujeres con adrenoleucodistrofia
Desde la década de 1980, se conoce que las mujeres con adrenoleucodistrofia pueden desarrollar síntomas. En un estudio realizado en el AMC en los Países Bajos, se demostró que con el aumento de la edad, la frecuencia de las mujeres con adrenoleucodistrofia que son sintomáticas aumenta (Figura 1). Más del 80% de las mujeres con adrenoleucodistrofia desarrollará signos de disfunción neurológica hacia la edad de 60 años.
 Figura 1: Correlación entre el estado sintomático y la edad en una cohorte de 46 mujeres con adrenoleucodistrofia. Las barras verdes indican el porcentaje de mujeres en cada grupo de edad que han desarrollado síntomas clínicos relacionados con la adrenoleucodistrofia. Los puntos negros muestran cada individuo de la cohorte, clasificado como sintomático o presintomático.
Figura 1: Correlación entre el estado sintomático y la edad en una cohorte de 46 mujeres con adrenoleucodistrofia. Las barras verdes indican el porcentaje de mujeres en cada grupo de edad que han desarrollado síntomas clínicos relacionados con la adrenoleucodistrofia. Los puntos negros muestran cada individuo de la cohorte, clasificado como sintomático o presintomático.
Es muy importante señalar que los síntomas en la infancia son extremadamente raros. Además, la enfermedad del cerebro en las mujeres mayores con afectación de la médula espinal es muy poco común. Los síntomas en las mujeres afectadas son principalmente debidos a anormalidades en la médula espinal y los nervios de las piernas, de la misma forma que en varones afectados con la forma de la médula espinal (mielopatía). Durante décadas, debilidad y espasticidad de las piernas, sensación anormal de las extremidades inferiores, y deterioro del control sobre la vejiga y el intestino pueden ir apareciendo. A diferencia de los hombres afectados, es muy poco probable que las mujeres desarrollen insuficiencia suprarrenal, aunque se ha descrito en el 1% en un grupo grande. Ninguna de las portadoras holandesas estudiadas presentó signos de disfunción adrenal.
No está claro lo que impide a las mujeres con adrenoleucodistrofia el desarrollo de la forma cerebral de la enfermedad. El gen de la adrenoleucodistrofia se encuentra en el cromosoma X (véase la página Datos sobre la ALD para más información). Normalmente, uno de los dos cromosomas X que cada mujer tiene en las células de sus tejidos se inactiva al azar. Se plantea la hipótesis, parcialmente apoyada por estudios de laboratorio, que en las mujeres que desarrollan la forma cerebral de la adrenoleucodistrofia los cromosomas X no se inactivan al azar, pero que por alguna razón el cromosoma X normal, el cuál no presenta la mutación en el gen ABCD1, es inactivado en todas las células. Esto da lugar a una situación similar a la de los niños con adrenoleucodistrofia cerebral.
Por qué se deben identificar las mujeres con adrenoleucodistrofia
Muchas mujeres con adrenoleucodistrofia con síntomas leves permanecen durante años sin ser identificadas. Esto puede dar lugar al retraso de un tratamiento específico para la espasticidad, el dolor en la parte baja de la espalda o en las articulaciones, y disfunción de la vejiga y el intestino. Una vez diagnosticadas, estos síntomas pueden ser tratados con mayor facilidad. Lo más importante es que una vez que una mujer es diagnosticada como heterocigota para una mutación de la adrenoleucodistrofia, sus hijos y parientes pueden ser examinados, con el fin de diagnosticar a niños y hombres con insuficiencia suprarrenal. Además, las pruebas prenatales (uso de líquido amniótico o biopsia de vellosidades coriónicas) permite la identificación temprana de un feto afectado. Los niños y hombres que se ven afectados y que tienen una disfunción todavía no reconocida de las glándulas suprarrenales pueden ser identificados. Si se deja sin tratamiento, la disfunción suprarrenal puede dar lugar a complicaciones graves e incluso la muerte. Los niños afectados por la enfermedad pueden ser monitoreados estrechamente; cuando desarrollan afectación cerebral, pueden someterse a un trasplante de médula ósea o de células madre. Esto ha demostrado ser beneficioso en los niños con afectación cerebral leve. A las hijas de las mujeres con adrenoleucodistrofia que desean tener hijos también se les puede ofrecer la posibilidad de realizarles las pruebas de ADN y el análisis de VLCFA.
Estableciendo el diagnóstico
La adrenoleucodistrofia solía ser diagnosticada mediante el análisis de VLCFA en muestras de sangre o células de la piel cultivadas (fibroblastos). En niños y hombres, un nivel anormalmente elevado de VLCFAs en el plasma sanguíneo o fibroblastos cultivados confirma el diagnóstico de ALD. Sin embargo, en al menos del 10 al 15% de las mujeres con adrenoleucodistrofia, los niveles de VLCFA están dentro de límites normales. Aparentemente, sus cromosomas X no afectados tienen suficiente actividad residual y son capaces de enmascarar la deficiencia bioquímica. Por lo tanto, es imposible excluir que una mujer en una familia afectada no tenga adrenoleucodistrofia al encontrar concentraciones normales de VLCFA en la sangre o células cultivadas! Por otra parte, una vez que se encuentran niveles elevados de VLCFA, se confirma el diagnóstico.
En la actualidad, el diagnóstico de ADN (análisis de mutaciones) es la prueba “golden standard” para la identificación de las mujeres que padecen de adrenoleucodistrofia o de las cuáles se sospecha que padecen esta enfermedad. Esto nunca debe ser omitido en las mujeres de aquellas familias afectadas en las que el análisis de VLCFA está dentro de los límites normales. Hasta el momento, se han identificado más de 800 mutaciones diferentes en el gen ABCD1. El análisis de ADN puede realizarse en sangre u otras muestras de tejido, incluyendo biopsias de vellosidades coriónicas y células del líquido amniótico, o las células cultivadas previamente.
Estrategia diagnóstica
Cuando la adrenoleucodistrofia se diagnostica en una familia, no se puede enfatizar lo suficiente en lo importante que es examinar al resto de la familia de forma exhaustiva. Esto puede prevenir nuevos casos innecesarios y ayuda a identificar a aquellos parientes con insuficiencia suprarrenal y a aquellos que pueden beneficiarse del trasplante de médula ósea o de células madre. Para hacer la identificación de los familiares afectados más fácil y fiable, no sólo el análisis de VLCFA y el análisis de ADN deberían llevarse a cabo una vez que la adrenoleucodistrofia se diagnostica en una familia. Una vez que se ha encontrado la mutación, el análisis mutacional en los familiares es mucho más fácil, ya que la prueba se puede centrar en esa anormalidad en específico. El análisis de ADN debe realizarse en centros especializados.
La disfunción hormonal
Aunque la disfunción endocrina se ha descrito en las mujeres con adrenoleucodistrofia, es muy poco común. Aproximadamente el 1% tiene algunos signos de deterioro de la función de las glándulas suprarrenales. La función anormal de la glándula tiroides ha sido descrita, pero se encuentra con frecuencia en la población en general, por lo que es difícil decir si existe alguna relación con la adrenoleucodistrofia. En general, las pruebas endocrinas sólo deberían llevarse a cabo cuando se sospeche de insuficiencia endocrina.
Neuroimagen y de electrofisiología
Sólo en raras ocasiones se han reportado graves anomalías de la materia blanca en las mujeres con adrenoleucodistrofia. Si se sospecha de afectación cerebral se puede confirmar con IRM. Exámenes electrofisiológicos, tales como el electroencefalograma (EEG) o pruebas de potenciales evocados, pueden ser anormales en estos casos también. Cuando los nervios periféricos están involucrados, los estudios de conducción nerviosa y la electromiografía (EMG) pueden ser anormales también.
Para fines de investigación estas pruebas son interesantes, pero no es esencial para llevarlas a cabo a menos que existan anomalías claras en el examen físico que requieran más investigación.
El tratamiento y la gestión
El tratamiento curativo
En las últimas décadas muchas terapias han sido probadas en la adrenoleucodistrofia. El tratamiento con aceite de Lorenzo, interferón ß, inmunoglobulinas intravenosas, inmunosupresión con fármacos citostáticos y el recambio plasmático hasta ahora no han sido capaces de detener o retrasar la progresión de la enfermedad una vez que los síntomas neurológicos se hallan presentes. Hasta ahora, el trasplante de médula ósea o células madre en los niños con afectación cerebral leve es el único tratamiento que ha demostrado ser beneficioso.
En los hombres y mujeres con síntomas relacionados con la mielopatía, sin embargo, el trasplante de médula ósea o de células madre no debe realizarse, ya que este tratamiento agresivo se dirige contra la inflamación cerebral. Esto no es efectivo frente a la pérdida lentamente progresiva de la función de las células nerviosas en la médula espinal (mielopatía) y los nervios periféricos, que se produce en las mujeres con adrenoleucodistrofia.
El tratamiento sintomático
La espasticidad se puede tratar mediante fármacos que reducen el tono muscular o, a veces con inyecciones de toxina botulínica en los músculos afectados.
Muchas mujeres con adrenoleucodistrofia experimentan dolor en la parte baja de la espalda o dolor en los tobillos, rodillas y caderas, causado por el aumento del tono muscular en las piernas y la consiguiente dificultad para caminar. Los analgésicos no opioides, tales como el acetaminofeno y los fármacos anti-inflamatorios no esteroideos (por ejemplo, ibuprofeno, naproxeno o diclofenaco) pueden ser particularmente útiles en el tratamiento del dolor.
Existen medicamentos disponibles para reducir los problemas de la incontinencia urgente, pero esto a menudo no será eficaz si los síntomas son graves. Dispositivos para el tratamiento de la incontinencia son a menudo necesarios.
Un enfoque multidisciplinar
Las mujeres con adrenoleucodistrofia con síntomas neurológicos pueden beneficiarse de un enfoque multidisciplinar. El neurólogo puede tratar algunos de los síntomas, pero derivarlas a un médico de rehabilitación o a un urólogo es a menudo necesario. Si es necesario, un psicólogo puede ser consultado para ayudar a lidiar con esto. Por último, pero no menos importante, el genetista clínico es esencial, dado que el modo de herencia de la adrenoleucodistrofia, la disponibilidad de pruebas prenatales, y la necesidad de la detección en la familia deben ser propuestas.
Observaciones finales
Muchas, si no la mayoría de las mujeres con adrenoleucodistrofia desarrollarán síntomas neurológicos causados por daños a la médula espinal y los nervios periféricos. Es importante reconocer estos síntomas para el tratamiento sintomático efectivo. Debe tenerse en cuenta que, en la actualidad, existen herramientas muy potentes y fiables de diagnóstico para demostrar o excluir del estado de portador de la adrenoleucodistrofia en mujeres. La prueba de oro para el diagnóstico en las mujeres es el análisis de ADN, ya que el análisis de VLCFA puede ser normal en el 10 al 15% de las mujeres con adrenoleucodistrofia. Una vez establecido el diagnóstico de la enfermedad, la familia debería ser examinada y los familiares varones afectados deberían ser identificados. Estas pruebas y el cribado familiar deben realizarse en centros especializados.
Adaptado y traducido con permiso de: van Geel BM. Draagsterschap van X-gebonden adrenoleukodystrofie (El estado de portador para la adrenoleucodistrofia ligada al X). Ned Tijdschr Geneeskd (2000) 144:1764-1768. Actualizado y combinado con nuevos datos de: Engelen et al., X-linked adrenoleukodystrophy in women: A cross-sectional cohort study. Brain. (2014) 137(Pt 3):693-706 (artículo gratuito).
Presentaciones clínicas
Authors: Stephan Kemp, Ph.D., Hugo Moser, M.D. and Marc Engelen, M.D., Ph.D
(Traducido por Nerea Montedeoca Vázquez (Biomedicine student))
Introducción
La Adrenoleucodistrofia ligada al X (ALD) es una enfermedad que se presenta de maneras variadas y diferentes (fenotipos). Los síntomas pueden ir desde una degeneración progresiva de la medula espinal en hombres y mujeres (mielopatía) hasta una enfermedad fatal a nivel cerebral en niños y hombres (ALD cerebral). La adrenoleucodistrofia es causada por un defecto genético (mutación) en el gen ABCD1. Todos los pacientes con adrenoleucodistrofia tienen una mutación en dicho gen. Más de 800 mutaciones diferentes han sido identificadas hasta el momento – la mayoría de ellas están recogidas en esta base de datos. Desgraciadamente las mutaciones no tienen un valor predictivo respecto a la severidad de la enfermedad en cada individuo afectado. Aunque todos los niños nacidos con adrenoleucodistrofia tienen una mutación en el gen ABCD1, el curso de la enfermedad en cada paciente resulta completamente impredecible, incluso en miembros de una misma familia que compartan la misma mutación. Por el momento no se ha identificado ningún marcador capaz de predecir la evolución de la enfermedad; ni los niveles en sangre de ácidos grasos de cadena muy larga (VLCFA), ni la mutación presente en el gen, ni la historia familiar respecto a la adrenoleucodistrofia, ni la presencia o ausencia de la proteína de la adrenoleucodistrofia en cultivos de líneas celulares.
Prevalencia de la ALD
La incidencia total de la ALD es uno de cada 14.000 nacidos vivos. La ALD ha sido identificada en todos los países europeos y latinoamericanos, China, Japón, India, Israel, Arabia-Saudí y en todos los grupos étnicos, incluidos los afroamericanos, nativos americanos y los Maoris.
Mujeres con ALD
Presintomáticas:La adrenoleucodistrofia es evidenciada mediante el cumplimiento de al menos uno de los siguientes criterios: evidencia de niveles elevados de ácidos grasos de cadena muy larga, mutación identificada en el gen ABCD1, ser hija de un afectado con adrenoleucodistrofia, o haber tenido al menos dos niños afectados por la adrenoleucodistrofia. El número de mujeres presintomáticas con adrenoleucodistrofia disminuye según aumenta su edad. La mayoría de mujeres menores de 30 años están libres de síntomas neurológicos.
Mielopatia: Como en muchas de las enfermedades ligadas al X, originalmente se pensaba que la mayoría de las mujeres con adrenoleucodistrofia estarían libres de síntomas. Sin embargo, en un estudio reciente se mostró que más del 80% de las mujeres con adrenoleucodistrofia desarrollan síntomas después de los 60 años. El texto completo de este estudio puede ser visualizado y descargado (como pdf). En general la aparición de síntomas neurológicos ocurre más tarde que en los hombres adultos afectados; normalmente entre los 40 y 50 años de edad (Fig 1). La discapacidad motora y la progresión de la enfermedad son generalmente menos severas pero algunas mujeres presentan síntomas tan severos como los hombres afectados. Es importante destacar que las mujeres con adrenoleucodistrofia son a menudo mal diagnosticadas con esclerosis múltiple. Aunque hay bastantes mujeres afectadas de adrenoleucodistrofia, la mayoría no están diagnosticadas puesto que muchas de ellas se mantienen asintomáticas hasta que alcanzan la mediana edad. Además, muchas mujeres con síntomas no son diagnosticadas a no ser que tengan un familiar varón afectado y sintomático. Un estudio del Instituto Kennedy Krieger demostró que de 616 familias identificadas con adrenoleucodistrofia, en solo 16 casos el primer paciente identificado en la familia había sido una mujer. La inmensa mayoría de mujeres con adrenoleucodistrofia solo son identificadas después de que se haya diagnosticado a un varón en la familia (por medio de programas de cribado extendidos a la familia). El mejor método para determinar si una mujer tiene adrenoleucodistrofia es a través de un análisis del gen ABCD1. No obstante, con tal de que este análisis sea más fiable, la mutación en esa familia debe ser identificada en un familiar varón afectado o bien en un portador obligado. Un portador obligado sería una mujer con un padre diagnosticado con adrenoleucodistrofia o una mujer con dos hijos, los cuales presenten un diagnóstico de adrenoleucodistrofia confirmado a través de pruebas genéticas.
Recomendamos que las mujeres en riesgo de presentar el gen afectado para la adrenoleucodistrofia sean informadas sobre la disponibilidad de pruebas genéticas cuando alcancen la edad reproductiva. En ese entonces, ellas podrán tomar una decisión informada respecto a su deseo de ser estudiadas y también se les ofrecerá la posibilidad de entrar en contacto con un asesor genético.
Afectación cerebral: Es raro encontrar mujeres con adrenoleucodistrofia y afectación cerebral pero algún caso aislado ha sido descrito.
Síntomas clínicos de insuficiencia suprarrenal primaria: La insuficiencia suprarrenal o enfermedad de Addison (ver más abajo) es muy rara en mujeres con adrenoleucodistrofia a cualquier edad, pues se presenta en menos del 1% de los casos.

Figura 1: Correlación entre el estatus de sintomática y la edad, en una cohorte de 46 mujeres con adrenoleucodistrofia. Las columnas verdes indican el porcentaje de mujeres distribuidas por grupos de edad en función de la aparición de síntomas relacionados con la adrenoleucodistrofia. Los círculos negros representan a cada individuo de la cohorte, clasificado como sintomático o presintomático.
Hombres con ALD
Pre-sintomáticos: Algunos pacientes, especialmente aquellos identificados por cribado familiar, pueden no presentar afectaciones neurológicas ni anomalías endocrinas. La adrenoleucodistrofia ha sido diagnosticada por uno de los siguientes criterios: demostración de niveles elevados de VLCFA o una mutación identificada en el gen ABCD1. Sin embargo, no hay evidencia de afectación suprarrenal o afectación neurológica todavía. El número de varones pre-sintomáticos disminuye con la edad. Es importante que un médico haga un seguimiento de estos pacientes con frecuencia. El seguimiento en niños y hombres pre-sintomáticos con adrenoleucodistrofia es importante por dos razones: 1) la detección precoz de la insuficiencia suprarrenal, y 2) la detección temprana de adrenoleucodistrofia cerebral por resonancia magnética del cerebro para poder proponer el trasplante de células madre hematopoyéticas (TCMH) si un donante HLA-compatible o un cordón umbilical se hayan disponibles. El riesgo de que estos pacientes desarrollen síntomas neurológicos o anomalías endocrinas es alta. Es común no presentar síntomas antes de los 3 primeros años de edad y muy raro después de los 40 años.
Insuficiencia suprarrenal: Una evaluación prospectiva de la función suprarrenal en 49 niños neurológicamente pre-sintomáticos con adrenoleucodistrofia mostró que el 80% ya tenía una alteración de la función suprarrenal. La insuficiencia suprarrenal no diagnosticada es conocida por ser una causa frecuente de morbilidad y puede prevenirse con un control cuidadoso, la identificación precoz de afectación de la reserva suprarrenal, y la iniciación oportuna de la terapia de reemplazo hormonal. A menudo, esta es la primera manifestación de la adrenoleucodistrofia, con frecuencia antes de la aparición de los síntomas neurológicos. Los signos más comunes de insuficiencia suprarrenal son crónicos o de larga duración, fatiga, debilidad muscular, pérdida del apetito, pérdida de peso, dolor abdominal y vómitos inexplicables. Otros síntomas pueden incluir náuseas, diarrea, presión arterial baja (que cae aún más cuando la persona se pone de pie, causando mareo o desmayo), irritabilidad y depresión, antojo de alimentos salados, hipoglucemia, o bajo nivel de azúcar en la sangre, dolor de cabeza, o sudoración. Los individuos pueden o no presentar aumento de la pigmentación de la piel que resulta de la secreción excesiva de la hormona corticotropina (ACTH).
Adrenoleucodistrofia cerebral (infancia, pubertad y edad adulta): Los síntomas de adrenoleucodistrofia cerebral progresan, en general, rápidamente. Los síntomas específicos dependen de la edad de presentación. La adrenoleucodistrofia cerebral ocurre a menudo en la infancia temprana (adrenoleucodistrofia cerebral infantil; CCALD), pero nunca antes de la edad de 3 años. Por lo general, los niños afectados inicialmente tienen problemas de comportamiento o déficits de aprendizaje, a menudo diagnosticados como trastorno por déficit de atención o hiperactividad. Estos comportamientos pueden persistir durante meses o más, pero luego son seguidos por síntomas que sugieren una enfermedad subyacente más grave. Estos síntomas pueden incluir “distraerse” en la escuela: falta de atención, deterioro de las habilidades de escritura, y disminución del rendimiento escolar; dificultad en la comprensión del habla (aunque la percepción del sonido es normal); dificultad en la lectura, la orientación espacial y la comprensión de material escrito; torpeza; alteraciones visuales y de vez en cuando doble visión; así como un comportamiento agresivo o desinhibido. La resonancia magnética cerebral en este momento puede ser sorprendentemente anormal incluso cuando los síntomas son relativamente leves. En algunos niños, las convulsiones pueden ser la primera manifestación. La tasa de progresión es variable. Puede ser extremadamente rápida con discapacidad total de seis meses a dos años, seguida de muerte a distintas edades. La mayoría de las personas ya sufren deterioro de la función suprarrenal en el momento en que se observan los primeros trastornos neurológicos. Un paciente varón recién nacido tiene un riesgo del 35-40% de desarrollar adrenoleucodistrofia cerebral entre las edades de 3 y 18 años, pero la adrenoleucodistrofia cerebral también puede ocurrir en la edad adulta. En pacientes adultos la presentación y la progresión de la adrenoleucodistrofia cerebral es similar, con cambios en el comportamiento y/o síntomas psiquiátricos. Los adultos que desarrollan adrenoleucodistrofia cerebral a menudo ya tienen signos de adrenomieloneuropatia (AMN) e insuficiencia suprarrenal.
Mielopatía: Prácticamente todos los pacientes con adrenoleucodistrofia que llegan a la edad adulta desarrollan afectación de la médula espinal (mielopatía). Los síntomas se limitan a la médula espinal y los nervios periféricos. El diagnóstico de la adrenoleucodistrofia rara vez se hace durante los primeros 3-5 años de síntomas clínicos, a menos que se hayan identificado otros casos de adrenoleucodistrofia dentro de la misma familia. El cuadro típico es el de un hombre de unos veinte años o de mediana edad el cual desarrolla una rigidez progresiva y debilidad en sus piernas, deterioro de la sensación de vibración en las extremidades inferiores, alteraciones del esfínter, y disfunción sexual. Todos los síntomas son progresivos durante décadas. El pelo de los pacientes con AMN es delgado y escaso; estos pacientes a menudo muestran calvicie a los veinte años de edad. La mayoría de los hombres con esta presentación clínica presentan deterioro de la función suprarrenal en el momento en que se observan los primeros síntomas neurológicos. La combinación de mielopatía y disfunción suprarrenal se denomina adrenomieloneuropatía (AMN). Los hombres con mielopatía también pueden desarrollar adrenoleucodistrofia cerebral. El riesgo exacto se desconoce y la aparición de la adrenoleucodistrofia cerebral no se puede predecir. El riesgo de desarrollar adrenoleucodistrofia cerebral secundaria a mielopatía se estima que es de al menos un 20% a lo largo de un período de 10 años.
Ácidos Grasos de Cadena Muy Larga
Desde el punto de vista bioquímico, ¿cuál es el problema en ALD / AMN?
En los pacientes con ALD y AMN, se encuentra una elevación de los niveles de acidos grasos de cadena muy larga, estos se acumulan en la mayoría de los tejidos del cuerpo. Este exceso es más grave en el cerebro y las glándulas suprarrenales y da lugar a problemas neurológicos y mal funcionamiento de la glándula suprarrenal (enfermedad de Addison). Los AGCML también se acumulan en el plasma sanguíneo. Esto hace que sea posible diagnosticar ALD por un análisis de sangre.
Niveles normales de AGCML en una mujer sospechosa de padecer ALD. ¿Qué significa ésto?
El análisis AGCML plasma es la prueba diagnóstica más utilizada para la ALD. El ensayo depende de la demostración de un aumento de los niveles de C26: 0 y el aumento de la C26: 0 / C22: 0 y C24: 0 / C22: 0 proporcionales. La experiencia con este ensayo en más de 2.000 pacientes con ALD (hombres y mujeres) ha demostrado que aproximadamente el 10 y el 20% de las mujeres con ALD tienen niveles normales de AGCML.
Niveles normales de acidos grasos de cadena muy larga no excluye el diagnóstico de la ALD. Cuando se sospecha que una mujer puede ser una posible portadora de ALD, el análisis de la mutación es el método más fiable para el diagnóstico. Lo ideal sería que la mutación en la familia fuera definida en un varón afectado u heterocigota olbligada (una portadora obligada es una mujer, ya sea con un padre diagnosticado con ALD o una mujer con dos niños con ALD demostrado genéticamente).
¿Que son los ácidos grasos?
Antes de que la estructura química de los ácidos grasos pueda ser explicada, se necesitan unos pocos conocimientos previos de química.
La mayoría de los productos químicos que nos son familiares, como la sal y el agua, se componen de diferentes módulos denominados “elementos”. La sal se compone de los elementos sodio (Na) y cloro (Cl). Su estructura química es muy simple: un átomo de sodio está vinculado a un átomo de cloro. La fuerza que los mantiene unidos se llama enlace químico. El sodio sólo puede unirse con un átomo de otro elemento, y lo mismo para el cloro. Por lo tanto, la estructura química de la sal puede ser escrita como Na-Cl. La línea que une a Na y Cl representa el enlace químico.
El caso del agua es un poco más complicado. Se compone de los elementos hidrógeno (H) y oxígeno (O). El hidrógeno es como el Na y Cl que sólo pueden unirse con otro elemento. El oxígeno puede formar dos enlaces. En el agua, un O se puede unir con dos H. Por lo tanto, podemos escribir la estructura del agua como HOH. El hecho de que los elementos pueden unirse con otros elementos ha permitido a la naturaleza producir un número increíble, casi ilimitado, de productos químicos que componen nuestro entorno y a nosotros mismos.
Los ácidos grasos están formados por hidrógeno (H), oxígeno (O) y carbono (C). El carbono puede formar cuatro enlaces, lo que lo hace un elemento muy versátil. La parte grasa de los ácidos grasos es una cadena de átomos de carbono unidos entre si y cada C está también unido a varios H.

La parte “ácida” de un ácido graso tiene un C, dos O y un H.
En la parte ácida, hay dos líneas o enlaces entre el C y uno de los O. Esto se conoce como “doble enlace”.

Ejemplos de ácidos grasos
El ácido acético (vinagre), un ácido graso de cadena corta; C2:0

El ácido palmítico (se encuentra en la grasa de cerdo o la manteca), es un ácido graso de cadena larga; C16:0

Dos ácidos grasos de cadena muy larga (AGCML), se denominan ácido lignocerico; C24:0

y ácido hexacosanoico; C26:0

Un sistema de abreviación conveniente se muestra en los ácidos grasos mencionados arriba. El número después de la “C” es el número de átomos de carbono en la cadena, por ejemplo, C2, C16, C24, C26, etc. El número después de los dos puntos nos dice el número de dobles enlaces en la cadena de carbono. En todos los ejemplos anteriores, no hay dobles enlaces entre los átomos de carbono, por lo tanto, todos tienen la designación “: 0”.
Ácidos grasos – ¿De qué se tratan? ¿De dónde vienen y qué hace el cuerpo con ellos?
Los ácidos grasos son productos químicos necesarios en el cuerpo. Se encuentran en los compuestos más complejos, como los triglicéridos, fosfolípidos, esfingolípidos, glucolípidos, y otros. Los triglicéridos, son los principales productos químicos en la “grasa”, son principalmente una forma de almacenamiento. Los fosfolípidos son parte de las membranas que rodean todas las células en el cuerpo y las estructuras más pequeñas dentro de las células. Esfingolípidos y glicolípidos son sustancias químicas complejas que se encuentran principalmente en las células cerebrales y nerviosas; los gangliósidos y la mielina se encuentran en esta categoría. Por lo tanto, los ácidos grasos son absolutamente necesarios para muchas funciones corporales normales. No podríamos vivir sin ellos.
Los ácidos grasos están presentes en los alimentos que comemos. Se encuentran en cantidades especialmente altas en los alimentos grasos, fritos, y los aceites. También están presentes en grandes cantidades en las nueces y semillas. La carne, aun los cortes magros, tienen una gran cantidad de ácidos grasos. Por otra parte, verduras, frutas y alimentos ricos en almidón (por ejemplo, pasta o pan) son relativamente bajos en ácidos grasos.
¿Qué sucede con los ácidos grasos de la dieta? Los ácidos grasos y otros nutrientes en los alimentos llegan al estómago donde comienzan el proceso de digestión. En este proceso, los alimentos se descomponen en sus diferentes componentes, tales como hidratos de carbono (azúcares y almidones), proteínas y lípidos (ácidos grasos y colesterol). Estos componentes son absorbidos por las células que recubren los intestinos. Los nutrientes pasan a través de las células intestinales y de los vasos sanguíneos pequeños. Estos vasos sanguíneos se dirigen directamente al hígado, el cual puede ser comparado con una “planta de procesamiento” de nutrientes. En las células del hígado, los nutrientes son metabolizados, lo que significa que son desglosados para producir calor o energía, o son convertidos en otros productos que el cuerpo necesita. El exceso de nutrientes puede ser convertido a una forma de almacenamiento como el glucógeno (para los azúcares) o los triglicéridos (para los ácidos grasos).
A menudo el cuerpo tiene un exceso de ácido grasos, ya sea porque se incorpora una gran cantidad con la dieta o porque se produce demasiado internamente. Por lo tanto, tenemos que deshacernos de dicho exceso. Los ácidos grasos son degradados u oxidados para producir energía o calor para el cuerpo. De hecho, son la principal fuente de combustible durante la inanición. Existe un delicado equilibrio entre tener suficiente nivel de ácidos grasos circulando y tener demasiado. El cuerpo normalmente tiene mecanismos para mantener este equilibrio. Cuando el equilibrio se desplaza, la enfermedad aparece.
VLCFA, ¿Qué sabemos acerca de ellos?
1) ¿Están presentes normalmente en el cuerpo?
– ¡Sí!
2) ¿Cuál es su función?
– Son parte de las membranas del cerebro, incluyendo la mielina, el “aislamiento” alrededor de las fibras nerviosas.
3) ¿De dónde vienen?
– De la dieta y a través de la elongación de los ácidos grasos más cortos.
4) ¿Qué ocasiona que aumenten los niveles de AGCML en ALD / AMN?
– El cuerpo produce demasiado.
– El cuerpo no elimina las cantidades en exceso
5) ¿Cuál de estas posibilidades es la correcta?
– Los estudios con pacientes voluntarios y en células en el laboratorio han demostrado que el proceso que normalmente rompe u oxida los AGCML es defectuoso en ALD / AMN.
¿Cómo hace el cuerpo normalmente para realizar la oxidación de los ácidos grasos?
A través de una serie de reacciones químicas, el cuerpo acorta los ácidos grasos mediante la eliminación de dos átomos de carbono a la vez:
Beta-oxidación de los ácidos grasos

Un ácido graso no es químicamente muy reactivo.
La enzima acil-CoA sintetasa une la Coenzima A a los ácidos grasos

(“ácidos grasos activados”)
La enzima acil-CoA oxidasa inserta un “doble enlace”

La enzima enoil-CoA hidratasa utiliza una molécula de agua para remover el doble enlace y agregar un grupo hidroxilo (O-H).

El grupo “O-H” es oxidado a “=O” por la enzima hidroxiacil-CoA dehidrogenasa.

El ácido graso de 16 carbonos es cortado por una enzima thiolasa en un ácido graso de 14 carbonos y un ácido graso de 2 carbonos.
 +
+ 
Este acído graso activado de 14 carbonos puede seguir siendo acortado por repetición del proceso anterior.
La unidad de 2 carbonos puede ser usada para la producción de energía.
¿Cómo trabaja la enzima?

Las enzimas son proteínas en el cuerpo que ayudan a hacer que las reacciones químicas se produzcan. “Las enzimas tienen sitios de unión” para los productos químicos con los que interactuan. En esta ilustración, se muestra la primera reacción química de oxidación de ácidos grasos. La enzima tiene un sitio de unión para un ácido graso y otro para una sustancia química llamada coenzima A (CoA). Estos sitios de unión son muy específicos; ácidos grasos y CoA encajan en ellos como llaves en las cerraduras. Al reunir estos dos compuestos químicos cerca uno del otro, la enzima les permite unirse entre sí, formando una nueva sustancia química llamada graso acil-CoA. Al producirse esta unión es entonces libre para unirse a otro ácido graso y CoA. Sin la enzima, la probabilidad de que el ácido graso y la CoA se unan es casi cero.
¿Qué es la proteina ALD y como funciona?

Las células contienen estructuras vesiculares pequeñas llamadas orgánulos. Esta compartimentación biológica permite que el cuerpo ponga las enzimas que necesitan trabajar juntas en el mismo lugar. La Oxidación de ácidos grasos tiene lugar dentro de dos orgánulos diferentes – las mitocondrias y los peroxisomas. Las enzimas necesarias para la oxidación de ácidos grasos en la mitocondria son diferentes a las de los peroxisomas. Las investigaciones realizadas en el Instituto Kennedy Krieger en la década de 1980 por los Dres. Inderjit Singh y Hugo Moser mostraron que la oxidación de VLCFA tiene lugar en los peroxisomas solamente (Singh et al 1981).
En 1993, los Dres. Patrick Aubourg y Jean-Louis Mandel en París descubrieron que el gen ALD genera una proteína (ALDP) que se localiza dentro de las células en la membrana de los peroxisomas (Mosser et al 1993). La proteína ALD pertenece a una familia específica de proteínas transportadoras. Estas proteínas permiten a moléculas atravesar las membranas biológicas, como las que rodean a los orgánulos celulares.
ALD se caracteriza por la incapacidad de las células para metabolizar / degradar VLCFA a los ácidos grasos de cadena más corta. Esto se traduce en niveles elevados AGCML en todos los tejidos del cuerpo. La degradación de AGCML se lleva a cabo exclusivamente en los peroxisomas. Las enzimas que son necesarias para la descomposición de AGCML son funcionales y presente el interior de los peroxisomas en pacientes ALD. ALDP transporta AGCML a través de la membrana peroxisomal. Los experimentos que usan células de levadura y células de pacientes ALD proporcionan evidencia de que de hecho ALDP transporta AGCML (como VLCFA-CoA) a través de la membrana peroxisomal (van Roermund et al 2008; Ofman et al 2010).
La deficiencia de ALDP en ALD tiene dos importantes consecuencias: 1) Se deteriora la beta-oxidación peroxisomal AGCML y 2) que eleva los niveles de AGCML-CoA en el citosol de la célula. Estos niveles elevados de AGCML-CoA en el citosol son sustrato para la elongación posterior a incluso más ácidos grasos por ELOVL1, la elongasa específica C26-humana (Ofman et al 2010; Kemp y Wanders 2010).
ALDP
La proteína ALD (ALDP) no es detectable por medio de análisis de inmunofluorescencia en aproximadamente 70% de los individuos afectados. Por razones que no se comprenden bien, el producto del gen puede estar ausente incluso en las personas que tienen mutaciones sin sentido. El principio anomalía bioquímica es la acumulación de ácidos grasos de cadena muy larga saturados, en particular hexacosanoico (C26: 0) y tetracosanoico (C24: 0), como resultado de la alteración en la capacidad para degradar estas sustancias, una función que normalmente se realiza en el peroxisoma. La proteína ALDP transporta AGCML desde el citosol a la peroxisomas.

Figura: Detección de ALDP mediante inmunofluorescencia. (Izquierda) fibroblastos de una tinción punteada control, que indica la presencia normal de ALDP en los peroxisomas. (Centro) fibroblastos de un paciente varón con ALD y una mutación que afecta a la estabilidad ALDP. Tenga en cuenta que no hay tinción punteada. (Derecha) fibroblastos de un paciente con ALD de la misma familia que el paciente masculino. El gen ABCD1 se encuentra en el cromosoma X. Las mujeres tienen dos cromosomas X. Sin embargo, en cada celda sólo 1 cromosoma X está activo. Las células que muestran tinción punteada son aquellos que tienen una copia activa del gen normal ABCD1, mientras que aquellos que no muestran tinción punteada son las células que tienen un gen activo ABCD1 portadoras de la mutación. Las imágenes fueron tomadas por el Dr. Merel Ebberink
Historia de ALD
1910: En retrospectiva, Haberfeld y Spieler presentó la primera descripción clínica de un paciente con la adrenoleucodistrofia ligada al cromosoma X (Haberfeld y Spieler, 1910). Un niño de 6 años previamente sano había desarrollado una piel profundamente bronceada (hiperpigmentación), deterioro de la agudeza visual, y su rendimiento escolar se deterioró. Los siguientes meses, apareció la incontinencia, perdió la capacidad de hablar y tetraparesia espástica, que con el tiempo evolucionó a una incapacidad para caminar. Fue hospitalizado a la edad de 7, 8 y murió meses después. Un hermano mayor había muerto de una enfermedad similar a la edad de 8. Examen histológico post-mortem del cerebro reveló extensos cambios en la materia blanca del cerebro, junto con la acumulación perivascular de linfocitos y células plasmáticas en el sistema nervioso, lo que indica una respuesta inflamatoria.
1923: Siemerling y la enfermedad de Creutzfeldt reportaron el caso de un niño con una progresión de la enfermedad similar, incluyendo la piel oscura y los hallazgos neuropatológicos como en el caso descrito por Haberfeld y Spieler en 1910, con la excepción de que la atrofia de la corteza suprarrenal fue documentada.
1963: Hasta el momento nueve casos similares se han informado. El hecho de que todos los pacientes eran varones sugirió ligada al cromosoma X como herencia recesiva (Fanconi et al 1963).
1970: El nombre adrenoleucodistrofia se introdujo basado en la asociación llamativa de una leucodistrofia con insuficiencia adrenocortical primaria (adrenal) (Blaw, 1970).
1972: La clave para todo el conocimiento posterior acerca de la enfermedad fue la observación hecha por Powers, Schaumburg, y Johnson que las células suprarrenales de pacientes ALD contenían inclusiones característica de lípidos (gotitas de grasa), seguido por la demostración de que estas gotitas de grasa consistió en ésteres de colesterol que contenía una llamativo y exceso de de ácidos graso de cadena muy larga (VLCFA, AGCML).
1976: Una forma adulta más lentamente progresiva de la enfermedad que se caracteriza por la insuficiencia suprarrenal, mielopatía y neuropatía periférica fue descrito (Budka et al 1976). Un año más tarde, otros cinco casos fueron reportados por Griffin et al. quien propuso esta presentación clínica de ALD a ser nombrado Adrenomieloneuropatía (AMN) (Griffin et al., 1977; Schaumburg et al. 1977).
1981: La identificación de VLCFA como un biomarcador para ALD condujo al desarrollo de una prueba diagnóstica para la ALD basada en la demostración de niveles elevados de VLCFA en células cultivadas de la piel (fibroblastos), plasma, glóbulos rojos y amniocitos (Moser et al. 1981). Estas pruebas han permitido diagnostico postnatal preciso y el diagnóstico prenatal. Estudios metabólicos demostraron que VLCFA se metabolizan (a través de la beta-oxidación) exclusivamente en orgánulos subcelulares llamadas peroxisomas y esta oxidación de VLCFA se reduce en fibroblastos de pacientes ALD (Singh et al 1981). Por lo tanto, ALD es una enfermedad peroxisomal.
1981: El locus ALD fue mapeado en el segmento terminal del brazo largo del cromosoma X, Xq28 (Migeon et al., 1981).
1982: El primer trasplante de médula ósea (TMO) se realizó en un niño con ALD cerebral. Un trasplante de médula ósea alogénico de un hermano donante HLA idéntico normales se realizó en un niño de 13 años de edad, con progresión rápida ALD. El injerto y la recuperación hematológica completa se produjo dentro de 4 semanas. Diez días después del trasplante de médula ósea, los niveles y la enzima AGCML actividad de glóbulos blancos volvió a la normalidad; después de 3 meses, hubo una reducción progresiva de plasma AGCML a niveles sólo ligeramente por encima de lo normal. Pero el deterioro neurológico continuó. El paciente murió de una infección por adenovirus 141 días después del trasplante de médula ósea.
1986: Rizzo et al. demostró que la adición de ácido oleico (C18: 1) al medio de cultivo de tejidos normaliza los niveles de VLCFA saturada en fibroblastos de piel cultivados de pacientes con ALD. Estos hallazgos fueron la base para el desarrollo de aceite de Lorenzo. El tratamiento de los pacientes con ALD con aceite de Lorenzo normaliza los niveles plasmáticos VLCFA el plazo de 4 semanas (Moser et al. 1987). Varios estudios abiertos han demostrado que el aceite de Lorenzo no pudo mejorar la función neurológica o endocrina ni tampoco detener la progresión de la enfermedad. Desafortunadamente, la eficacia clínica de aceite de Lorenzo nunca ha sido evaluado en un ensayo clínico controlado con placebo adecuado. En 2001, el profesor Hugo Moser escribió: “Es nuestra opinión de que la terapia de aceite de Lorenzo no está justificada en la mayoría de los pacientes que ya tienen síntomas neurológicos. El beneficio clínico de aceite de la vida es limitado en el mejor de los casos”.
1990: El equipo del Prof. Patrick Aubourg informó sobre el primer trasplante de médula ósea (TMO) (Aubourg et al 1990). Se había trasplantado un niño de 8 años de edad con leve afectación neurológica, neuropsicológica leve y anomalías leves de resonancia magnética. Su afectado gemelo no idéntico fue el donante. El paciente se recuperó por completo y la neurológica, neuropsicológica y las anormalidades de IRM desapareció. Cuando se realiza en la etapa más temprana de la desmielinización cerebral, un trasplante de células de médula ósea o de progenitores hematopoyéticos (TPH) puede estabilizar o incluso revertir la desmielinización cerebral en los niños o adolescentes con ALD.
1993: Un equipo dirigido por los Dres. Mandel y Aubourg identificaron el gen de la ALD (ABCD1) utilizando estrategias de clonación posicional (Mosser et al. 1993). La identificación del gen ALD permite la detección de mutaciones causantes de enfermedades, diagnóstico prenatal y la prueba de portador.
1997: Tres laboratorios informaron de la generación de un modelo de ratón para ALD (Forss-Petter et al., 1997; Kobayashi et al., 1997; Lu et al., 1997). Mientras que el ratón ALD exhibe las mismas anormalidades bioquímicas como se observa en los pacientes, el ratón no desarrolla ALD (Pujol et al., 2001).
1999: La base de datos de ALD fue creada por Hugo Moser y Stephan Kemp. Inicialmente sólo sirvió como un registro de todas las mutaciones identificadas en el gen ABCD1, pero poco después fue ampliado para proporcionar información sobre muchos aspectos de la ALD.
2001: Se informó y estableció que la ALD afecta a todos los grupos étnicos y es el trastorno peroxisomal más común con una incidencia estimada de 1: 17.000 (hombres y mujeres combinados) (Bezman et al., 2001). Esto hace que la ALD sea la leucodistrofia hereditaria más común.
2005: Bioquímicamente, ALD no sólo se caracteriza por un defecto en la degradación de VLCFA en los peroxisomas, sino que también hay un aumento en la posterior cadena-elongación de VLCFA (Kemp et al. 2005).
2006: El equipo dirigido por el Dr. Ann Moser desarrolló un método de análisis de alto rendimiento AGCML (con C26: 0-lisoPC como el metabolito de diagnóstico) para ser utilizado en muestras de sangre seca (Hubbard et al., 2006). Estos avances en la detección AGCML permitirán la adición de ALD a los programas de cribado neonatal.
2009: Un equipo dirigido por los Dres. Cartier y Aubourg informaron sobre el éxito del tratamiento de dos niños de 7 años de edad con signos tempranos de ALD cerebral y requiere terapia génica (Cartier et al., 2009). Las imágenes por resonancia magnética del cerebro y las pruebas cognitivas mostraron que la progresión de la enfermedad cerebral paró de 14-16 meses después del tratamiento. Esto es comparable con el resultado clínico de TPH.
2010: El equipo de investigación del Dr. Stephan Kemp estableció que transporta ALDP AGCML través de la membrana peroxisomal. Una deficiencia en ALDP tiene dos efectos principales: por un lado, perjudica la degradación peroxisomal de VLCFA, y por el otro lado, aumenta los niveles citosólicos de VLCFA. el C26 elongasa específica humana (Ofman et al. 2010).
2014: En los Estados Unidos, el Estado de Nueva York comenzó el cribado neonatal para la ALD (Vogel et al 2015). El diagnóstico precoz de la ALD es la clave para salvar vidas, ya que el cribado neonatal permite la monitorización prospectiva y la intervención temprana.
2015: En los Estados Unidos, Connecticut inició el cribado neonatal de ALD. En Europa, los Países Bajos amplió su programa nacional de cribado neonatal de 17 a 31 condiciones, incluyendo la ALD.
2016: El 16 de febrero, ALD se incluye en las pruebas de cribado neonatal en los Estados Unidos Recomendado Grupo Proyección Uniforme (RUSP). En los Estados Unidos, California inició el cribado neonatal de ALD.