
Tatsachen zur ALD
Definition
Die Adrenoleukodystrophie (ALD) ist eine ernste fortschreitende genetische Krankheit, die die Nebennieren, das Rückenmark und die weiße Substanz des Nervensystems schädigt. Sie wurde erstmals 1923 erkannt und anfangs Schilder-Krankheit oder sudanophilie Leukodystrophie genannt. Im Jahre 1970 wurde die Bezeichnung Adrenoleukodystrophie geprägt. „Adreno“ bezieht sich auf die Nebennieren (glandulae adrenales); “leuko” auf die weiße Substanz des Gehirns, und „Dystrophie“ bedeutet Störung von Wachstum oder Entwicklung. Die Krankheit hat nichts zu tun mit der neonatalen Adrenoleukodystrophie, die zu den Störungen des Zellweger-Spektrums gehört.
Biochemie
Die ALD ist eine erbliche Stoffwechselkrankheit, bei der ein Fehler in einem spezifischen Enzymsystem zur Anhäufung von sehr langkettigen Fettsäuren (Very Long Chain Fatty Acids, VLCFA) in allen Geweben des Körpers führt. Diese besonderen Fettsäuren (VLCFA) schädigen Zellen und Gewebe. Aus noch ungeklärten Gründen sind vor allem das Gehirn, das Rückenmark, die Hoden und die Nebennieren betroffen. Im Zentralnervensystem führt die Anhäufung der VLCFA schließlich zur Zerstörung der Markscheiden (des Myelins), welche die Nervenfasern umgeben und zu entsprechenden neurologischen Störungen. Die VLCFA schädigen auch die Zellen der Nebennieren und verursachen dadurch die Addison-Krankheit (Nebennierenversagen).

Abbildung 1: Die sehr langkettigen Fettsäuren (VLCFA), die sich bei der ALD anhäufen, entstehen in den Zellen hauptsächlich durch Verlängerung von langkettigen Fettsäuren. Um die vorhandene Menge von VLCFA sorgfältig im Gleichgewicht zu halten, müssen VLCFA auch wieder abgebaut werden. Die VLCFA können nur in Peroxisomen abgebaut werden. Peroxisomen gibt es in allen Zellen des Körpers, außer den roten Blutkörperchen. Die ALD entsteht durch Mutationen im ABCD1-Gen, das für die Herstellung des ALD-Proteins (ALDP) verantwortlich ist. Das ALDP funktioniert als Transporter für VLCFA aus dem Cytosol in das Peroxisom. Innerhalb der Peroxisomen sind zwar alle Enzyme vorhanden, die VLCFA abbauen können, aber die VLCFA können nicht dorthin gelangen.
Epidemiologie
Die ALD kommt überall auf der Welt vor und ist nicht auf bestimmte ethnische Gruppen und Regionen beschränkt. Die Gesamtinzidenz der ALD beträgt etwa 1 auf 15.000 Neugeborene.
Genetik
Die ALD ist eine geschlechtsgebunden vererbte Krankheit, was bedeutet, dass das ALD-Gen (seine offizielle Bezeichnung ist ABCD1) auf dem X-Chromosom liegt. Männer haben ein X-Chromosom und ein Y-Chromosom (XY; Abbildung 2). Wenn der Vater das fehlerhafte ALD-Gen trägt, gibt es kein zweites, schützendes X-Chromosom; daher bekommt er ALD. Frauen haben zwei X-Chromosomen (XX; Abbildung 2). Frauen, die das fehlerhafte Gen tragen, wurden üblicherweise als „Überträgerinnen“ bezeichnet, weil man dachte, dass nur ein geringer Anteil solcher Frauen Krankheitssymptome bekäme. Inzwischen wurde jedoch klar, dass dies nicht der Fall ist (see below and the Women with ALD page). Die Symptome bei Frauen sind etwas milder als bei Männern, doch bei 80% der Frauen mit ALD treten schließlich Symptome auf. Der Ausdruck „ALD-Überträgerin“ ist daher irreführend und sollte nicht mehr verwendet werden. Wahrscheinlichste Erklärung dafür, dass Frauen eine mildere Form der Erkrankung bekommen, ist das Vorhandensein eines normalen ABCD1-Gens auf ihrem anderen X-Chromosom. Man nimmt an, dass das Vorhandensein von Zellen mit einem gesunden ABCD1-Gen Frauen mit ALD vor dem Auftreten der Gehirn-Form der ALD (zerebrale ALD) schützt.

Abbildung 2 (Links): Wenn eine Frau Überträgerin für ALD ist, sind die Aussichten für jedes Neugeborene wie folgt: wenn das Kind eine Tochter ist, ist die Wahrscheinlichkeit 50%, dass die Tochter Überträgerin für ALD ist und 50%, dass sie nicht betroffen ist. Wenn das Kind ein Junge ist, ist die Wahrscheinlichkeit 50%, dass der Junge ALD hat und 50%, dass er nicht betroffen ist. (Rechts): Wenn ein Mann mit einer geschlechtsgebunden vererbten Krankheit wie ALD Kinder hat, dann sind alle seine Söhne normal (er vererbt sein Y-Chromosom immer an seinen Sohn), aber alle seine Tochter sind Überträgerinnen (er vererbt sein einziges, fehlerhaftes X-Chromosom an seine Tochter).
Klinischer Verlauf
Patienten mit ALD zeigen bei Geburt keinerlei Symptome. Bei Knaben besteht die erste Manifestation gewöhnlich in einem Nebennierenversagen, das schon im Säuglingsalter auftreten kann. Im Erwachsenenalter tritt beim männlichen Geschlecht eine Myelopathie (Krankheit des Rückenmarks) auf. Männliche ALD-Patienten können eine fortschreitende zerebrale Demyelinisierung (zerebrale ALD) bekommen, sowohl im Kindes- als auch im Erwachsenenalter. Die zerebrale ALD kann Erstmanifestation der ALD sein oder zusätzlich zu Nebennierenversagen und/oder Myelopathie auftreten (Abbildung 3). Frauen mit ALD sind ebenfalls betroffen und nicht bloße Überträgerinnen des ALD-Gen-Defekts, da mehr als 80 % von ihnen im Alter von 60 Jahren Beschwerden und Symptome entwickeln, die mit einer Myelopathie zusammenhängen. Nebennierenversagen und zerebrale Demyelinisierung treten dagegen bei Frauen mit ALD nur selten auf.

Abbildung 3: Das klinische Spektrum der ALD im männlichen Geschlecht. Patienten mit ALD zeigen bei Geburt keine Symptome. Die farbigen Balken geben den Altersbereich für den Beginn des Nebennierenversagens (blauer Balken), der Myelopathie (rosa Balken) und der zerebralen ALD (grauer Balken) an. Das Nebennierenversagen kann schon im Alter von 5 Monaten beginnen. Im Erwachsenenalter tritt bei Männern stets eine chronisch progressive Myelopathie auf. Eine zerebrale ALD kann in jedem Alter auftreten, beim jüngsten bekannten Patienten war dies im Alter von 3 Jahren. Der primäre Defekt im ALD-Gen und die Ansammlung von VLCFA (sehr langkettigen Fettsäuren) in Geweben führen zum Nebennierenversagen und zur Myelopathie (was zusammen als Adrenomyeloneuropathie oder AMN bezeichnet wird). Die zerebrale ALD wird höchstwahrscheinlich in Gang gesetzt durch das Zusammenspiel des primären ALD-Gen-Defekts mit einer Kombination von noch unbekannten Auslösern aus der Umwelt und/oder genetischen Faktoren. Es ist wichtig zu wissen, dass bei Patienten mit Nebennierenversagen und/oder Myelopathie immer ein Risiko für das Auftreten der zerebralen ALD besteht.
Nebennierenversagen (oder sogar eine lebensbedrohliche Addison-Krise) kann das zuerst auftretende Symptom der ALD bei Knaben und Männern sein, Jahre oder gar Jahrzehnte vor dem Beginn neurologischer Symptome. Eine Studie bei neurologisch präsymptomatischen Jungen mit ALD zeigte, dass 80 % von ihnen schon zum Zeitpunkt der ALD-Diagnose eine beeinträchtigte Funktion der Nebennieren aufwiesen. Die häufigsten Anzeichen von Nebennierenversagen sind chronische oder langdauernde Müdigkeitszustände, Muskelschwäche, fehlender Appetit, Gewichtsverlust, Bauchschmerzen und unerklärliches Abbrechen. Zu weiteren Symptomen gehören Übelkeit, Durchfall, niedriger Blutdruck (der beim Aufstehen einer Person noch weiter fällt), Reizbarkeit und Depression, ein Bedürfnis nach salzigen Nahrungsmitteln, niedriger Blutzucker, Kopfweh oder Schweißausbruch. Betroffene Personen können (aber müssen nicht) eine verstärkte Hautpigmentierung aufweisen, die auf übermäßiger Sekretion von adrenocorticotropem Hormon (ACTH) beruht.
Myelopathie: Praktisch alle männlichen Patienten mit ALD, die das Erwachsenenalter erreichen, entwickeln eine Myelopathie, typischerweise zwischen dem 20. und 40. Lebensjahr. Die Symptome sind auf das Rückenmark und die peripheren Nerven begrenzt. Anfangs schreitet die neurologische Störung langsam voran. Die Diagnose ALD wird während der ersten 3-5 Jahre nach Auftreten der neurologischen Symptome nur selten gestellt, es sei denn in der Familie wurden bereits andere Fälle von ALD festgestellt. Bei den Patienten entsteht eine langsam voranschreitende Gangstörung infolge von Steifigkeit und Schwäche der Beine. Auch Blasensfunktionsstörungen können auftreten, mit Harndrang und Fortschreiten bis zur völligen Inkontinenz. Alle Symptome schreiten über Jahre oder Jahrzehnte voran, wobei bei den meisten Patienten das Gehen ohne Hilfe im fünften bis sechsten Lebensjahrzehnt verloren geht.
Adrenomyeloneuropathie (AMN): die Bezeichnung AMN bezieht sich auf männliche Patienten mit der Kombination von Nebennierenversagen und Myelopathie.
Zerebrale ALD: Bei Jungen und Männern mit ALD besteht das Risiko des Auftretens von Myelinschäden in der weißen Substanz des Gehirns (zerebrale ALD). Der Beginn einer zerebralen ALD wurde nie vor dem Alter von 3 Jahren beobachtet. Im Adoleszentenalter wurde die zerebrale ALD mit 4-7 % und im Erwachsenenalter mit 2-5 % bisher als seltenes Ereignis angesehen. Heute jedoch, wo wir systematisch eine große Gruppe von Männern mit ALD anhand von jährlichen MRT-Bildern verfolgen, scheinen die Zahlen in Wirklichkeit höher zu liegen. Ein möglicher umweltbedingter Auslöser ist ein Kopftrauma, doch werden wahrscheinlich noch andere – bisher unbekannte – genetische und Umweltfaktoren für das Entstehen einer zerebralen ALD benötigt. Die Symptome der zerebralen ALD sind im allgemeinen rasch voranschreitend. Das Risiko eines neugeborenen männlichen Patienten für das Auftreten einer zerebralen ALD beträgt im Alter von 13-18 Jahren 35-40 %. Bei Jungen im Grundschulalter sind die ersten Symptome meist Verhaltensstörungen und Lernschwierigkeiten, die sich im Abfall der schulischen Leistungen widerspiegeln. Diese frühen Symptome werden oft anderen Störungen wie dem Aufmerksamkeitsdefizit-Hyperaktivität-Syndrom zugeschrieben, wodurch die Diagnose der ALD verzögert werden kann. Bei erwachsenen Patienten sind die ersten Symptome oft ebenfalls psychiatrischer Art und können nach einer Depression oder Psychose aussehen. Bei diesen Patienten wird die Diagnose ALD oft verzögert, besonders wenn keine Familienvorgeschichte darauf hinweist und keine Symptome des Nebennierenversagens vorliegen. Nach Fortschreiten des Krankheitsprozesses werden neurologische Störungen offensichtlich, wozu Hör- und Sehstörungen, Schwäche in Armen und Beinen, Koordinationsstörungen und Krämpfe gehören. In diesem Stadium schreitet die Krankheit extrem rasch und völlig zerstörerisch voran. Die Patienten können innerhalb weniger Monate die Fähigkeit verlieren, Sprache zu verstehen und zu gehen. Schließlich werden sie bettlägerig, blind, unfähig zu sprechen oder zu antworten und benötigen Vollpflege und Fütterung mittels Nasen-Magen-Sonde oder durch Gastrostomie. Der Tod tritt im allgemeinen 2-4 Jahre nach dem Beginn der zerebralen Symptome ein, gelegentlich verbleiben gut gepflegte Patienten jahrelang in einem anscheinend vegetativen Zustand.
Frauen mit ALD: Wie bei manchen X-chromosomalen Krankheiten wurde ursprünglich angenommen, dass weibliche Personen, die das fehlerhafte ALD-Gen tragen, asymptomatisch bleiben. Inzwischen ist jedoch klar geworden, dass diese Ansicht nicht stimmt. Tatsächlich bekommen mehr als 80 % der Frauen mit ALD im Alter von 60 Jahren Symptome (siehe Seite „Frauen mit ALD“). Die vollständige Veröffentlichung mit der Beschreibung von Befunden und Symptomen bei Frauen mit ALD kann eingesehen und heruntergeladen werden (als PDF). Im allgemeinen treten bei Frauen neurologische Symptome später auf als bei Männern mit Myelopathie; typischerweise geschieht dies im Alter von 40-50 Jahren. Das voranschreiten der Krankheit ist in der Regel langsamer als bei Männern. Bemerkenswerterweise, und im Gegensatz zu Männern, ist Stuhlinkontinenz eine häufige Klage von Frauen mit ALD. Es muss festgehalten werden, dass die Myelopathie bei Frauen mit ALD oft als Multiple Sklerose verkannt wird. Sowohl Nebennierenversagen als auch zerebrale ALD sind bei Frauen sehr selten und betreffen jeweils weniger als ein Prozent. (Für mehr Einzelheiten siehe die Seite “Women with ALD”.).
Labortests
ALD wird durch einen einfachen Bluttest diagnostiziert, bei dem die sehr langkettigen Fettsäuren (VLCFA) gemessen werden. Dieser Test ist beim männlichen Geschlecht zuverlässig und allgemein akzeptiert als hochakkurate Methode der ALD-Diagnose bei allen Altersstufen des männlichen Geschlechts. Beim weiblichen Geschlecht zeigt dieser Test jedoch in 20 % normale Blutspiegel der der VLCFA und kann somit zu einem „falsch negativen“ Ergebnis führen. In solchen Fällen bringt ein DNA-Test klare Ergebnisse. Dieser Test erlaubt die korrekte Identifizierung von Frauen mit ALD durch den Nachweis von Mutationen im ALD-Gen, und ein normales Resultat dieser Untersuchung verschafft einer Frau Gewissheit, dass sie nicht Überträgerin des fehlerhaften ALD-Gens ist.
Neugeborenenscreening
Eine frühe Diagnose der ALD kann lebensrettend sein, weil das Neugeborenenscreening prospektiv das Auftreten des Nebennierenversagens und der zerebralen ALD erfassen lässt. Ein Test für das Neugeborenenscreening ist entwickelt worden. Mit ihm lässt sich ein erhöhter Gehalt von VLCFA (in Form von C26:0-Lysophosphatidylcholin) in Trockenblutproben feststellen. Am 30. Dezember 2013 hat der Staat New York damit ein Neugeborenenscreening auf ALD begonnen. Im Februar 2016 wurde die ALD auf die offizielle Liste der in den Vereinigten Staaten für das Neugeborenenscreening empfohlenen Krankheiten gesetzt. Seither haben weitere US-Staaten und andere Länder mit Screeningprogrammen begonnen oder haben Prozesse in Gang gesetzt, um die ALD ihren bisher vorhandenen Screeningprogrammen hinzuzufügen. Einzelheiten und letzte Informationen über das Neugeborenenscreening auf ALD finden sich auf der Seite „Neugeborenenscreening“.
Forschung
Über ALD wird auf der ganzen Welt intensiv geforscht. Im Jahre 1993 wurde das Gen für ALD durch gemeinsame Anstrengung der Ärzte Patrick Aubourg und Jean-Louis Mandel in Frankreich und Hugo Moser in USA identifiziert. Dies hat Türen geöffnet zu weiteren Studien. Die Forschungsaktivitäten verfolgen zahlreiche Richtungen, um grundlegende Fragen zu klären wie etwa: „Auf welche Weise führen erhöhte VLCFA zur Zerstörung von Myelin?“oder „Warum erkrankt ein Patient an zerebraler ALD, während ein anderer (bei dem es sich sogar um den Bruder des Patienten handeln kann) später im Leben eine Myelopathie bekommt?“.
Behandlung
Derzeit gibt es keine heilende Behandlung für ALD.
Hormonersatztherapie mit Nebennierensteroiden: die meisten männlichen ALD-Patienten bekommen ein Nebennierenversagen. Nebennierenversagen ist oft das erste Anzeichen einer ALD: eine aufschlussreiche Studie zeigte, dass bei 80 % von Jungen mit ALD, die noch ohne neurologische Symptome waren und durch ausgedehnte Familienuntersuchungen identifiziert worden waren, im Alter von vier Jahren ein Nebennierenversagen bestand. Für diese Patienten ist die Hormonersatztherapie mit Nebennierensteroiden unerlässlich und kann lebensrettend sein, doch hat die erfolgreiche Therapie des Nebennierenversagens keine Auswirkungen auf die neurologischen Symptome.
Für die Myelopathie, die 85 % aller ALD-Patienten (beide Geschlechter zusammen genommen) befällt, gibt es keine heilende Behandlung.
Diätbehandlung: Weil die VLCFA toxisch für das Myelin, die Nebennieren und die Hoden sind, wurde verschiedentlich versucht die Konzentration der VLCFA im Blutplasma zu senken. Die verminderte Aufnahme von VLCFA mit dem Essen hat allein keinen Effekt auf die VLCFA-Spiegel im Plasma.
Lorenzos Öl: VLCFA werden vorwiegend durch Kettenverlängerung kürzerer Fettsäuren synthetisiert. Im Labor führt die Zugabe von einfach ungesättigten Fettsäuren zum Zellkulturmedium von ALD-Fibroblasten zur Normalisierung der VLCFA-Konzentrationen. Man erklärt dies damit, dass die für die Synthese der VLCFA benötigten Enzyme für einfach ungesättigte Fettsäuren dieselben sind wie für gesättigte Fettsäuren. Die Affinität dieser Enzyme zu den einfach ungesättigten Fettsäuren ist jedoch höher. Aus dieser Beobachtung ergab sich die Grundlage eines diätetische Behandlungsversuchs. Die orale Verabreichung von Ölsäure in Form von Triglyceriden (GTO) und von Erucasäure in Form von Triglyceriden (GTE) normalisierte die VLCFA-Plasmaspiegel bei den meisten Patienten mit ALD innerhalb eines Monats. Die Kombination von GTO und GTB im Verhältnis 4:1 wurde als Lorenzos Öl bekannt, als Tribut für Lorenzo Odone, den ersten mit dieser Mischung behandelten Patienten. Von Lorenzos Öl versprach man sich viel. Verschiedene nicht verblindete (open label) Versuchsreihen haben jedoch gezeigt, dass das Öl nicht in der Lage war die neurologischen oder hormonellen Funktionsstörungen zu bessern oder das Fortschreiten der Krankheit zum Stillstand zu bringen (zu mehr Einzelheiten siehe die Seite Lorenzos Öl).
Lovastatin: Von dieser Substanz wurde ein Effekt auf die VLCFA gezeigt, doch konnte das Ergebnis von anderen Gruppen nicht reproduziert werden. Tatsächlich zeigten spätere Experimente, dass Statine keinen günstigen Effekt auf die VLCFA in Gehirn und Nebennieren von ALD-Mäusen hatten und sogar die VLCFA in diesen Geweben erhöhten. Wegen dieser sich widersprechenden Ergebnisse wurde im Academic Medical Center in Amsterdam eine randomisierte doppelblinde und placebokontrollierte klinische Studie durchgeführt, um die Wirkung von Lovastatin als Mittel zur Absenkung der VLCFA bei ALD zu testen. Die Ergebnisse und Schlussfolgerungen sprachen dafür, dass die Behandlung mit Lovastatin zu einem geringen Absinken der VLCFA-Spiegel im Plasma führte, aber auf zellulärer Ebene keinen Effekt auf die VLCFA hatte, da der Gehalt an der Fettsäure C26:0 in roten und weißen Blutkörperchen unverändert blieb. (Zu Einzelheiten siehe die Seite Lovastatin).
Bezafibrat: Auf der Suche nach Verbindungen, die möglicherweise VLCFA-Spiegel erniedrigen können, wurde Bezafibrat, ein für die Behandlung von Hyperlipidämien verwendetes Medikament, als VLCFA-absenkende Substanz erkannt. Versuche mit Fibroblasten zeigten, dass Bezafibrat deren VLCFA-Gehalt erniedrigte durch direkte Hemmung der Aktivität des VLCFA-spezifischen kettenverlängernden Enzyms, der Elongase ELOVL1. Eine unverblindete Pilotstudie wurde durchgeführt um den Effekt von Bezafibrat auf die Anhäufung von VLCFA in Blutkörperchen von AMN-Patienten zu beurteilen. Unglücklicherweise war Bezafibrat nicht in der Lage die VLCFA-Spiegel in Blutkörperchen dieser Patienten zu senken. Höchstwahrscheinlich beruht dies darauf, dass mit diesem Medikament bei den Patienten keine ausreichenden Spiegel der Substanz erzeugt werden konnten.
Knochenmarktransplantation: Bei Jungen und Adoleszenten im Frühstadium einer zerebralen ALD kann eine allogeneische hämatopoetische Stammzelltransplantation (HSCT) das Voranschreiten der zerebralen Demyelinisierung stoppen, vorausgesetzt diese Maßnahme wird in einem sehr frühen Stadium der zerebralen Erkrankung durchgeführt. Die Wirksamkeit der HSCT beruht auf dem Ersatz von ALDP-defizienten Mikrogliazellen des Gehirns durch normale Mikrogliazellen, die aus Stammzellen des Knochenmarkspenders hervorgehen. (zu Einzelheiten siehe die Seite hämatopoetische Stammzelltransplantation, HSCT).
Gentherapie: Voraussichtlich wird in nicht zu ferner Zukunft die Transplantation von autologen hämatopoetischen Zellen (vom Patienten selbst stammende Knochenmarkzellen), die mittels eines lentiviralen Vektors genetisch korrigiert wurden und dem Patienten wieder infundiert werden, eine zusätzliche therapeutische Option sein. Diese Einschätzung beruht auf den berichteten höchst ermutigenden Ergebnissen bei den ersten beiden so behandelten Patienten. (Zu Einzelheiten siehe die Seite Gentherapie bei ALD).
Eine Zusammenfassung zur ALD (10 Minuten)
Produced by Youreka Science in collaboration with ALD Connect, Inc.
Please see the ALD Connect Educational Videos & Webinars page for more videos
Sehr langkettige Fettsäuren
Biochemisch betrachtet, was ist das Problem bei ALD/AMN?
Bei Patienten mit ALD und AMN sammeln sich in den meisten Geweben des Körpers zu viele sehr langkettige Fettsäuren an (Very Long Chain Fatty Acids, VLCFA). Die übermäßige Ansammlung ist in Gehirn und Nebennieren am stärksten ausgeprägt und führt einerseits zu neurologischen Problemen, andererseits zu einer Funktionsstörungen der Nebennieren (Addison-Krankheit). Sehr langkettige Fettsäuren sind auch im Blutplasma erhöht. Dies macht die Diagnose der ALD durch eine Blutuntersuchung möglich.
Normaler Gehalt an VLCFA bei einer Frau mit Verdacht auf ALD. Was bedeutet dies?
Die Untersuchung von Blutplasma auf sehr langkettigen Fettsäuren (VLCFA) ist der am häufigsten verwandte diagnostische Test für ALD. Der Test besteht im Nachweis von erhöhten Spiegeln der Fettsäure C:26:0 und einem erhöhten Verhältnis der Fettsäuren C26:0/C22:0 sowie C24:0/C22:0. Erfahrungen mit diesem Text bei mehr als 2000 X-ALD-Patienten haben gezeigt, dass ungefähr 10-20% obligater Überträgerinnen (Frauen mit ALD) normale Blutspiegel der VLCFA haben.
Ein normaler Blutspiegel von VLCFA schließt daher den Überträgerstatus für ALD nicht aus. Wenn eine weibliche Person im Verdacht steht, Überträger für ALD zu sein, ist die Mutationsanalyse der sicherste Weg für die Identifizierung von Heterozygoten (Überträgerinnen). Idealerweise muss die Mutation in einer Familie bei einer erkrankten männlichen Personen oder bei einer obligaten heterozygoten Verwandten festgestellt worden sein.
Was sind Fettsäuren?
Um die chemische Struktur von Fettsäuren zu erklären braucht man etwas chemisches Hintergrundwissen.
Die meisten chemischen Stoffe mit denen wir vertraut sind, wie etwas Salz und Wasser, bestehen aus verschiedenen Bausteinen, die „Elemente“ genannt werden. Salz besteht aus den Elementen Natrium (Na) und Chlor (Cl). Seine chemische Struktur ist sehr einfach: ein Atom oder Molekül Natrium ist mit einem Atom Chlor verbunden. Die Kraft, die beide zusammen hält, nennt man chemische Bindung. Natrium kann nur eine Bindung mit einem anderen Molekül haben; dasselbe gilt für Chlor. Deshalb kann man die chemische Struktur von Salz schreiben als Na-Cl. Der Strich zwischen Na und Cl stellt die chemische Bindung dar.
Wasser ist etwas komplizierter. Es besteht aus den Elementen Wasserstoff (Hydrogenium, H) und Sauerstoff (Oxygenium, O). Wasserstoff kann, sowie Natrium und Chlor, nur eine Bindung mit einem anderen Element haben. Sauerstoff kann zwei Bindungen ausbilden. Bei Wasser verbinden sich zwei H mit einem O. Daher können wir die Struktur von Wasser schreiben als H-O-H. Die Tatsache, dass Elemente Bindungen mit anderen Elementen eingehen können, hat der Natur die Bildung einer unglaublichen, fast grenzenlosen Anzahl von chemischen Stoffen ermöglicht, aus denen wir und unsere Umwelt bestehen.
Fettsäuren bestehen aus H, O und Kohlenstoff (C). Kohlenstoff kann vier Bindungen ausbilden; dies macht Kohlenstoff zu einem sehr vielseitigen Element. Der Fett-Anteil einer Fettsäure ist eine Kette von miteinander verbundenen Kohlenstoffatomen; dabei ist jedes C mit einigen H’s verbunden.

Der “Säure“-Anteil einer Fettsäure hat ein C, zwei O’s und ein H.
Im sauren Anteil des Moleküls gibt es zwei Linien oder Bindungen zwischen dem C und einem der O’s. Dies nennt man „Doppelbindung“.

Beispiele von Fettsäuren
Essigsäure, eine kurzkettige Fettsäure; C2:0

Palmitinsäure (in Speck und Butter enthalten) ist eine langkettige Fettsäure; C16:0

Zwei sehr langkettige Fettsäuren (VLCFA) heißen Tetracosansäure; C24:0

und Hexacosansäure; C26:0

Für jede Fettsäure ist hier auch ein bequemes Abkürzungssystem angegeben. Die Zahl nach dem “C“ ist die Anzahl der Kohlenstoffatome in der Kette, z.B. C2, C16, C24, C26, usw.. Die Zahl nach dem Doppelpunkt nennt uns die Anzahl der Doppelbindungen in der Kohlenstoffkette. Bei den obigen Beispielen gibt es keine Doppelbindungen zwischen dem Kohlenstoffatomen; deshalb haben alle die Bezeichnung „:0“.
Fettsäuren – wozu sind Sie gut? Wo kommen Sie her und was macht der Körper damit?
Fettsäuren sind für den Körper notwendige Stoffe. Man findet sie in komplexer gebauten chemischen Stoffen wie Triglyzeriden, Phospholipiden, Sphingolipiden und anderen. Triglyzeride sind die Hauptbestandteile von „Fett“; sie sind in erster Linie eine Speicherform. Phospholipiden sind Bestandteile der Membranen, die alle Zellen im Körper umgeben und auch kleinerer Strukturen im Inneren der Zellen. Sphingolipide und Glykolipide sind komplexe chemische Stoffe, die sich hauptsächlich in Gehirn und der Nervenzellen finden; Ganglioside und Myelin gehören in diese Gruppe. Fazit: Fettsäuren sind absolut notwendig für viele Funktionen unseres Körpers. Ohne sie könnten wir nicht leben.
Fettsäuren finden wir in unseren Nahrungsmitteln. Einen hohen Anteil davon haben fettige Nahrungsbestandteile, Frittiertes und Öle. Auch Nüsse und Samen enthalten viel davon. Fleisch, sogar magere Wurst, enthalten eine Menge Fettsäuren. Auf der anderen Seite sind Gemüse, Früchte und stärkehaltige Nahrungsmittel (zum Beispie Nnudeln oder Brot) verhältnismäßig arm an Fettsäuren.
Was passiert mit den gegessenen Fettsäuren? Fettsäuren und andere Nahrungsbestandteile kommen in den Magen, wo der Verdauungsprozess beginnt. Bei diesem Vorgang wird die Nahrung in verschiedene kleinere Bestandteile aufgespalten, wie Kohlenhydrate (Zucker und Stärke), Eiweiße (Proteine), und Lipide (Fettsäuren und Cholesterin). Diese Bestandteile der Nahrung werden dann von den Zellen der Darmwand aufgenommen. Nahrungsstoffe wandern durch die Datenzellen in kleine Blutgefäße. Diese Blutgefäße gehen direkt zur Leber, die man sich als Fabrik zur Verarbeitung von Nahrungsstoffen vorstellen kann. In den Leberzellen werden die Nahrungsstoffe metabolisiert (umgewandelt); das bedeutet sie werden entweder abgebaut um Hitze oder Energie zu produzieren, oder sie werden in andere Stoffe umgewandelt, die der Körper braucht. Dafür nicht benötigte Nahrungsstoffe können in eine Speicherform umgewandelt werden wie Glykogen (für Zucker) oder Triglyzeride (für Fettsäuren).
Sehr oft hat der Körper mit zuviel Fettsäuren zu tun, entweder weil wir zu viel gegessen haben oder bei der Körper selbst zu viel davon produziert. Wir müssen Fettsäuren loswerden können. Fettsäuren werden dann abgebaut oder “oxidiert” um Energie oder Wärme für den Körper zu bilden. In der Tat sind Fettsäuren die hauptsächliche Brennstoffquelle des Körpers im Hungerzustand. Es besteht ein heikles Gleichgewicht zwischen genügend Fettsäuren und zu viel davon. Normalerweise verfügt der Körper über einen fein eingestellten Mechanismus, um dieses Gleichgewicht aufrechtzuerhalten. Wenn dies nicht gelingt, entsteht oft eine Krankheit.
Sehr langkettige Fettsäuren (VLCFA) – was wissen wir darüber?
1) Kommen Sie normalerweise im Körper vor?
– JA!
2) Was ist Ihre Aufgabe?
– Sie sind Bestandteile von Membranen im Gehirn, auch von Myelin, der Isolationsschicht um die Nervenfasern.
3) Oder kommen Sie?
– Aus Nahrungsbestandteilen und durch Verlängerung kürzerer Fettsäuren im Körper.
4) Was könnte die Ursache des erhöhten Gehalts an VLCFA bei ALD/AMN sein?
– Der Körper stellt zu viel davon her.
– Der Körper baut nicht genügend davon ab.
5) Welche dieser beiden Möglichkeiten ist richtig?
– Studien bei freiwilligen Patienten und an Zellen im Labor haben gezeigt, dass der normale Prozess des Abbaus oder der Oxidation von VLCFA bei ALD/AMN defekt ist.
Wie oxidiert der Körper normalerweise Fettsäuren?
In einer Reihenfolge chemischer Reaktionen verkürzt der Körper die Fettsäuren dadurch, dass schrittweise jeweils zwei Kohlenstoffatome entfernt werden:
Beta-Oxidation der Fettsäuren

Eine Fettsäure ist normalerweise chemisch nicht sehr reaktiv.
Das Enzym Fettsäure-CoA-Synthetase bringt ein Coenzym A an die Fettsäure heran.

(“aktivierte Fettsäure“)
Das Enzym Acyl-CoA-Oxidase fügt eine „Doppelbindung“ ein.

Das EnzymEnoyl-CoA-Hydratase benutzt ein Wassermolekül um die Doppelbindung zu entfernen und ein Hydroxyl (O-H) einzuführen.

Das “O-H“ wird oxidiert zu “=O“ durch das Enzym Hydroxyacyl-CoA-Dehydrogenase.

TDie 16-Kohlenstoff-Fettsäure wird durch das Enzym Thiolase gespalten in eine 14-Kohlenstoff-Fettsäure und eine 2-Kohlenstoff-Fettsäure.
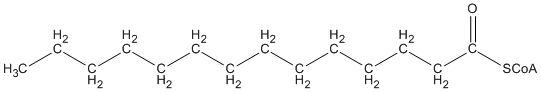 +
+ 
Die 14-Kohlenstoff-Fettsäure mit CoA-Gruppe kann durch Wiederholung desselben Prozesses weiter verkürzt werden.
Die 2-Kohlenstoff-Einheit kann zur Energieproduktion verwendet werden.
Wie arbeiten Enzyme?

Enzyme sind Eiweißstoffe des Körpers, die chemische Reaktionen in Gang bringen können. Im im Enzyme haben „Bindungsstellen“ für die Stoffe, mit denen sie in Wechselwirkung treten. Auf dem obigen Bild wird die erste chemische Reaktion der Fettsäure-Oxidation gezeigt. Das Enzym hat eine Verbindungsstelle für Fettsäuren und eine andere für einen Stoff, der Coenzym A (CoA) genannt wird. Diese Bindungsstellen sind sehr spezifisch; Fettsäuren und CoA passen da hinein wie Schlüssel ins Schloss. Indem das Enzym die beiden chemischen Stoffe nahe zueinander bringt, ermöglicht es ihnen eine Bindung einzugehen, wobei ein neuer Stoff gebildet wird, der Fettsäure-CoA (Acyl-CoA) genannt wird. Das Enzym lässt das Produkt los und ist dann wieder frei eine neue Fettsäure und neues CoA zu binden. Ohne das Enzym wäre die Wahrscheinlichkeit, dass Fettsäure und CoA eine Bindung eingehen, praktisch null.
Was ist das ALD-Protein und was tut es?

Zellen enthalten kleinere bläschenartige Strukturen, die Organellen genannt werden. Die dadurch mögliche räumliche Aufteilung innerhalb der Zellen erlaubt es dem Körper, diejenigen Enzyme zusammenzubringen, die zusammenarbeiten sollen. Oxidation von Fettsäuren findet in zwei verschiedenen Organellen statt – den Mitochondrien und den Peroxisomen. Die für die Fettsäurenoxidation in Mitochondrien benötigten Enzyme unterscheiden sich von denen in den Peroxisomen. Forschungsarbeiten von Dr. Inderjit Singh und Dr. Hugo Moser in den achtziger Jahren am Kennedy Krieger Institute haben gezeigt, dass die Oxidation der VLCFA nur in Peroxisomen stattfindet.
Im Jahre 1993 entdeckten Dr. Patrick Aubourg und Dr. Jean-Louis Mandel in Paris, dass das ALD-Gen ein Protein (ALD-Protein, ALDP) entstehen lässt, das in den Peroxisomen von Zellen lokalisiert ist. Das ALDP gehört zu einer spezifischen Familie von Transporterproteinen. Diese Proteine machen es Molekülen möglich, biologische Membranen zu durchqueren, wie diejenigen um Zellorganellen herum. ALDP wirkt wie ein Tor in der Membran, die das Peroxisom umgibt. Die VLCFA gehen durch das Tor und gelangen so ins Peroxisom, wo sie zu kürzeren Fettsäuren abgebaut werden.
Die wahrscheinlichste Erklärung für das, was bei der ALD nicht funktioniert, ist, dass im Zytoplasma der Zellen wartende VLCFA nicht ins Peroxisom gelangen können, weil das für den Transport durch die Membran zuständige Protein (ALDP) nicht richtig arbeitet.
Neugeborenenscreening
Einleitung
Babys mit ALD sind bei Geburt neurologisch normal. Bei Knaben kann jedoch eine frühe Diagnose zu lebensrettenden Maßnahmen führen. Zu diesen gehört einerseits der rechtzeitige Beginn einer Behandlung mit Nebennierenhormonen bei Nachweis des Versagens der Nebennieren, andererseits werden dadurch die Voraussetzungen für eine hämatopoetische Stammzelltransplantation (HSCT) geschaffen zur Behandlung einer zerebralen ALD. Die HSCT kann das oft tödliche Voranschreiten der zerebralen Demyelination stoppen, vorausgesetzt die Behandlung wird in einem sehr frühen Stadium der Krankheit durchgeführt. Leider ist diese Behandlung nur innerhalb eines engen Zeitfensters wirksam, das oft verpasst wird. Das Neugeborenenscreening ermöglicht die Nutzung dieses „Fensters des Möglichen“ durch rechtzeitige Einleitung dieser erprobten Therapien.
Im Februar 2016 wurde die ALD in den USA auf die Liste der für das Neugeborenenscreening empfohlenen Krankheiten gesetzt, d.h. auf eine bundesstaatliche Liste aller genetischen Krankheiten, die für die Programme des Neugeborenenscreenings in den Einzelstaaten empfohlen werden. Der Staat New York begann das Neugeborenenscreening für ALD am 30. Dezember 2013. Anschließend haben andere Staaten ebenfalls mit dem Screening für ALD begonnen (Abb. 1). In einigen anderen Staaten der USA sind die gesetzgeberischen Voraussetzungen für das Neugeborenenscreening auf ALD geschaffen worden. Außerhalb der USA hat in den Niederlanden der Gesundheitsminister zugestimmt, das Programms des Neugeborenenscreenings um ALD zu erweitern. Man geht davon aus, dass es Neugeborenenscreening für ALD in den genannten Staaten und Ländern beginnen wird, sobald Fragen der Finanzierung, der Testverfahren und die Regelung der Nachsorge geklärt sind.

Abbildung 1: Die Landkarte zeigt diejenigen Staaten der USA, wo das Neugeborenenscreening für ALD begonnen hat.
Kriterien für den Einschluss in das Screeningsprogramm
International besteht breite Übereinstimmung über die Kriterien für den Einschluss einer Krankheit in das Programm des Neugeborenenscreenings.
- Die frühe Diagnose muss einen unmittelbaren Vorteil für das Neugeborene haben. Der gesundheitliche Gewinn muss erheblich sein und als Ergebnis einer frühen Intervention bei einer schweren Krankheit mit bekanntem natürlichen Verlauf eintreten.
- Der Screeningtest muss von guter Qualität sein. Er muss eine hohe Spezifität und Sensitivität aufweisen, d.h. der Anteil sowohl falsch positiver als auch falsch negativer Ergebnisse muss sehr niedrig sein.
 Historisches
Historisches
Im Jahre 2004 hat Professor Hugo Moser bei einem Treffen des nationalen Beratungskomitees für das Neugeborenenscreening in USA vorgeschlagen, ALD auf die Liste der für das Neugeborenenscreening empfohlenen Krankheiten zu setzen. Das einzige Problem bestand darin, dass es einen verlässlichen Test für das Neugeborenenscreening noch nicht gab. Zur Überwindung des Problems sammelte er Geld und rekrutierte ein Team von Forschern am Kennedy-Krieger-Institut (Baltimore, MD), um einen geeigneten Biomarker zu finden und einen Test mittels Tandem-Massenspektrometertrie (MS/MS) zu entwickeln. Im Jahr 2006 berichtete das Team über die Identifizierung von C26:0-Lysophosphatidylcholin (C26:0-LPC) in Trockenblutproben, die nach der Geburt aus Venenblut von Knaben mit ALD gewonnenen waren (Hubbard et al. 2006). In den nachfolgenden Jahren haben Wissenschaftler die Analysetechnik weiter verbessert (Hubbard et al. 2009; Theda et al. 2014). Zusammen mit Forschern an der Mayo-Klinik (Rochester, Minnesota) wurde dann eine massentaugliche Methode für die Analyse des C26:0-LPC entwickelt (Haynes and De Jesús 2012; Turgeon et al. 2015). Im Jahre 2013 wurde diese Methode an 100.000 anonymen Trockenblutproben validiert.
 Das Aidan-Gesetz (Aidan’s Law)
Das Aidan-Gesetz (Aidan’s Law)
Im April 2012, nach dem Tod ihres Sohnes Aidan, der an cerebraler ALD gelitten hatte und zu spät diagnostiziert worden war, entwarf die Familie Seeger das Aidan-Gesetz und unterstützte seinen Weg in die Gesetzgebung des Staates New York. Das Gesetz wurde im Februar 2013 verabschiedet und trat im März 2013 in Kraft. Am 30. Dezember 2013 begann das Labor für Neugeborenenscreening des Staates New York mit dem Testen von Babys auf ALD.
Erste Erfahrungen im Staat New York
Während der ersten drei Jahre testete der Staat New York über 700.000 Neugeborene und identifizierte 45 Babys mit ALD: 22 Knaben und 23 Mädchen. Von diesen Zahlen ausgehend beträgt die Inzidenz der ALD bei Geburt 1 auf 15.000. Wenn ein Neugeborenes mit ALD identifiziert worden ist, wird der Arzt der Familie benachrichtigt und eine Überweisung an einen klinischen Genetiker veranlasst, um die Diagnose zu bestätigen. Außerdem erfolgt eine genetische Beratung und Unterstützung auf der Suche nach anderen Familienangehörigen, bei denen ein Risiko für ALD besteht (erweitertes Familienscreening).
Für Knaben ist es notwendig, planmäßig und wiederholt MRT-Untersuchungen des Gehirns durchzuführen, um die frühesten Anzeichen des Beginns einer zerebralen ALD zu erkennen. Außerdem müssen Tests der Funktion der Nebennieren veranlasst werden, um ein Versagen dieses Organs zu erkennen. Erforderlich ist eine umfassende Beurteilung neurologischer, neuropsychologischer und neuroradiologischer Befunde, sowie von Tests der Nebennierenfunktion, weil es keinen Test gibt, mit dem der klinische Verlauf eines einzelnen Babys mit einer ALD-Mutation vorausgesagt werden kann.
Der ALD-Test des Neugeborenenscreenings
Beim C26:0-LPC-Test und den dazu benötigten Materialien kann es in verschiedenen Laboratorien kleine Unterschiede geben, jedoch wird überall die ALD-Diagnose nach einem 3-Stufen-Schema gestellt (Abb. 2). Die erste Stufe besteht in einer massentauglichen Standard-MS/MS-Analyse von C26:0-LPC. Proben mit einem erhöhten Gehalt an C26:0-LPC werden dann in einer zweiten Stufe mittels HPLC-MS/MS analysiert. Diese Methode ist spezifischer, aber auch zeitaufwendiger. In solchen Proben, bei denen jetzt C26:0-LPC immer noch erhöht ist, findet als dritte Stufe die Sequenzierung des ABCD1-Gens statt.

Abbildung 2: Prinzip des 3-Stufen-Screenings auf ALD.
Erfordernisse
In verschiedenen Ländern gibt es maßgebliche Erfordernisse und laufende ethische Diskussionen im Hinblick auf die Einführung des Neugeborenenscreenings auf ALD.
- Das erste Kriterium für den Einschluss einer Krankheit in das Neugeborenenscreening, nach welchem die frühe Diagnose dem Neugeborenen einen unmittelbaren Vorteil bringen muss, kann ethische Besorgnisse auslösen. Bei ALD wird etwa ein Drittel der Knaben im Alter zwischen 3 und 18 Jahren eine zerebrale ALD entwickeln. Die übrigen zwei Drittel männlicher Patienten werden jedoch als Erwachsene eine Adrenomyeloneuropathie (AMN) bekommen, die sich durch Gliederspastik, Gangstörungen und Inkontinenz bemerkbar macht. Die AMN wird symptomatisch behandelt. Das Fehlen geeigneter Labortests oder anderer biologischer Merkmale macht die Voraussage des Krankheitsverlaufs bei einzelnen Patienten schwierig und kann daher das Risiko unnötiger medizinischer Interventionen in sich bergen.
- Das Neugeborenenscreening identifiziert auch Mädchen, die ein fehlerhaftes ALD-Gen tragen. Frauen mit ALD haben ein Risiko weit unter 1 % für das Auftreten eines Versagens der Nebennieren oder für eine zerebrale ALD. Ein neugeborenes Mädchen mit ALD hat daher keinen unmittelbaren Vorteil für seine Gesundheit, da eine Behandlung mit HSCT oder Nebennierenhormonen nicht infrage kommt. Etwa 80 % von Frauen mit ALD entwickeln im Alter von 60 Jahren eine Myelopathie.
- In verschiedenen Ländern wird unter Wissenschaftlern und innerhalb von Patientenorganisationen diskutiert, ob gewisse unbehandelbare Krankheiten in Programme des Neugeborenenscreenings aufgenommen werden sollten. Wie oben erwähnt, soll eine frühe Diagnose letztlich einen direkten Vorteil für die Gesundheit des Neugeborenen bringen. Dies kann im Falle einer diagnostizierten, aber nicht behandelbaren Krankheit nicht unmittelbar ersichtlich sein.
- Manchmal werden durch das Neugeborenenscreening Krankheiten identifiziert, die außerhalb des Zieles eines Tests liegen. Der Test des Neugeborenenscreenings auf C26:0-LPC identifiziert auch unbehandelbare Krankheiten die ebenfalls mit einem erhöhten Gehalt an C26:0-LPC einhergehen (Abb. 2). Zu diesen gehören die Krankheiten des Zellweger-Spektrums, die Störungen der peroxisomalen Fettsäureoxidation, die durch einen Defekt entweder der peroxisomalen Acyl-CoA-Oxidase 1 (ACOX1) oder des multifunktionalen Proteins (HSD17B4) verursacht werden, acyl-CoA binding domain containing protein 5 (ACBD5) deficiency, das “contiguous ABCD1 DXS1357E deletion syndrome” (CADDS), sowie das Aicardi Goutières Syndrome.
- Je nach Szenario können auch andere Vorteile jenseits solcher, die eindeutig auf die Verbesserung der Gesundheit des Neugeborenen abzielen, in Betracht gezogen werden. Einige davon können von Vorteil für das Neugeborene sein, etwa der schnellere Prozess einer Diagnostik. Die meisten Vorteile aber sind von klarem Nutzen für die Familie: die Möglichkeit eines erweiterten Familienscreenings, um zusätzliche Familienmitglieder mit einem ALD-Risiko zu identifizieren, und Anpassungen im Leben der Familie, um die Folgen der Krankheit in den Griff zu bekommen. Außerdem können Eltern von der Information durch ein Screening auf eine nicht wirksam behandelbare genetische Krankheit bei der Entscheidung über ihr weiteres Reproduktionsverhalten profitieren. Es bestehen jedoch auch klare Nachteile. Die Diagnose einer unbehandelbaren Krankheit kann ein Stigma sein und einen Schatten auf die Kindheit des Neugeborenen werfen.
Als Ergebnis der Tatsache, dass ALD in den USA auf die Liste der für das Neugeborenenscreening empfohlenen Krankheiten gesetzt worden ist, ist zu erwarten, dass das Neugeborenenscreening auf ALD in den nächsten Jahren in einer wachsenden Zahl von US-Staaten begonnen werden wird und dass andere Länder folgen werden. Diese Maßnahmen werden den klinischen Verlauf von hunderten von ALD betroffenen Babys beträchtlich verbessern und außerdem von Vorteil für ihre biologischen Verwandten und anderen Angehörigen sein.
Genetik und genetische Beratung
Identifizierung des ABCD1-Gens
Genetisches Linkage mit Glucose-6-Phosphat-Dehydrogenase (G6PD) ließ den ALD-Locus am Ende des langen Arms des X-Chromosoms in der Region Xq28 vermuten. Im Jahre 1993 wurde das X-ALD-Gen (ABCD1) mittels der Strategie des Positional Cloning identifiziert. Das ABCD1-Gen ist ungefähr 20 kb lang und enthält 10 Exons.
Das ALD-Protein (ALDP) gehört zur ABC-Superfamilie (ABC steht für ATP-Bindungs-Cassetten) von Transmembran-Transport-Proteinen. Das Protein besteht aus 745 Aminosäuren und enthält eine Membrandomäne mit sechs Transmembransegmenten in der Amino-Hälfte und einer ATP-Bindungs-Cassette in der Carboxy-Hälfte des Proteins. Immunzytochemische Studien haben gezeigt, dass es sich bei ALDP um ein peroxisomales Membranprotein handelt. Dies stimmt überein mit der bei ALD-Patienten beobachteten biochemischen Abnormität (defekte peroxisomale beta-Oxidation gesättigter sehr langkettiger Fettsäuren (VLCFA).
Mutationen wurden bei allen gründlich untersuchten ALD-Patienten gefunden. Komplementationsstudien an Fibroblasten, die von ALD-Patienten stammten, haben gezeigt, dass Expression des normalen ABCD1-Gens in Patientenzellen den VLCFA-Stoffwechsel normalisiert. Die stabile Expression von ALDP in ALD-Fibroblasten korrigiert auch den Gehalt an VLCFA auf ein normales Maß.
Die Funktion des ALDP und seine Rolle bei der Verstoffwechslung der VLCFA wurde kürzlich aufgeklärt. ALDP transportiert VLCFA-CoA-Ester durch die peroxisomale Membran. Das Fehlen von ALDP behindert den Transport von VLCFA und hat zwei Folgen: 1) eine herabgesetzte peroxisomale beta-Oxidation von VLCFA und 2) einen erhöhten Gehalt von VLCFA-CoA-Estern im Cytosol, die ein Substrat für weitere Kettenverlängerung zu längeren Fettsäuren darstellen.
Immunozytochemische Studien haben gezeigt, dass bei 70% aller ALD-Patienten das ALDP mittels Immunfluoreszenz nicht nachgewiesen werden kann. Alle Mutationen, abgesehen von Missense-Mutationen, beeinträchtigen die Stabilität von ALDP.
Genetische Beratung
ALD wird X-chromosomal rezessiv vererbt. Das ABCD1-Gen ist das einzige Gen, dass mit ALD assoziiert ist.
Alle Töchter einer von ALD betroffenen männlichen Person sind Überträgerinnen; keiner ihrer Söhne ist betroffen. Eine Überträgerin hat bei jeder Schwangerschaft ein 50%iges Risiko, die ABCD1-Mutation weiterzugeben. Söhne, die die Mutation erben, werden befallen sein; Töchter, die die Mutation erben, sind Überträgerinnen.
Der Bereich der phänotypischen Expression bei ALD und die Prognose einer betroffenen männlichen Person ist in unvorhersehbarer Weise variabel und kann NICHT vorhergesagt werden aufgrund von Blutspiegeln der VLCFA oder gezüchteter Hautfibroblasten, der Residualaktivität der beta-Oxidation in ALD-Hautfibroblasten, der Familienvorgeschichte oder der Art der Mutation des ABCD1-Gens, die beim Patienten identifiziert wurde. Dieselbe Mutation kann mit jedem der bekannten klinischen Phänotypen assoziiert sein. Milde Phänotypen können mit großen Deletionen assoziiert sein, die zu einem Totalausfall des Genprodukts führen, und schwere Phänotypen kommen bei Missense-Mutationen vor, bei denen ALDP in normaler Menge produziert wird. Weit auseinanderklaffende klinische Phänotypen kommen oft innerhalb einer einzelnen Sippschaft oder Geschwisterreihe vor. Die häufigste ABCD1-Mutation, eine Deletion von zwei Basenpaaren in Exon 5, wurde in Zusammenahng mit allen ALD-Phänotypen beobachtet. Die Gruppe von Dr. Johannes Berger hat eine Familie identifiziert, in der sechs betroffene Mitglieder fünf verschiedene Phänotypen aufwiesen.
Segregationsanalysen legen nahe, dass die phänotypische Variabilität auf einem autosomalen Modifier-Gen beruht. Umweltfaktoren könnten jedoch ebenfalls daran beteiligt sein, worauf die phänotypische Variabilität bei einem monozygoten Zwillingspaar hinweist.
Für Risikopaare ist es wichtig sich klarzumachen, dass sich weit voneinander unterscheidende Phänotypen oft innerhalb derselben Sippe oder Geschwisterreihe finden. So muss man Familien, die relative milde Phänotypen erlebt haben, darauf hinweisen, dass betroffener Nachwuchs einen schweren Phänotyp aufweisen kann.

Vererbungsmodus
Geschwister eines Probanden: Das Risiko von Geschwistern hängt vom genetischen Status der Eltern ab, der durch Stammbaumanalyse, Messung der VLCFA oder molekulargenetische Analyse geklärt werden kann.
Eltern eines männlichen Probanden oder einer weiblichen Probandin: Etwa 93% der Indexfälle haben die ABCD1-Mutation von einem Elternteil geerbt; 7% der Individuen mit ALD haben eine de novo Mutation.
Es ist sinnvoll, bei den Müttern sowohl von betroffenen männlichen Patienten als auch von Überträgerinnen, und bei den Vätern von Überträgerinnen die Blutspiegel der VLCFA zu bestimmen. Wenn die krankheitsverursachende Mutation bei einem betroffenen Familienmitglied identifiziert wurde, kann die Mutationsanalyse des ABCD1-Gens zur Beurteilung der Eltern verwendet werden.
Wenn die Mutter Überträgerin ist, ist das Risiko der Übertragung der krankheitsverursachenden Mutation bei jeder Schwangerschaft 50%. Männliche Geschwister, die die Mutation geerbt haben, werden befallen sein; weibliche Geschwister, die die Mutation geerbt haben, werden Überträgerin sein.
Wenn der Vater des Probanden eine krankheitsverursachende Mutation des ABCD1-Gens hat, sind alle weiblichen Geschwister Überträgerinnen, während keiner der männlichen Geschwister betroffen ist.
Wenn keiner der Eltern Überträger ist, ist das Risiko von Geschwistern eines Probanden gering.
Nachwuchs eines Probanden: Betroffene männliche Personen übertragen die ABCD1-Mutation auf alle ihre Töchter und keinen ihrer Söhne.
Überträgerinnen haben bei jeder Schwangerschaft ein 50%iges Risiko, die ABCD1-Mutation weiterzugeben. Söhne, die die Mutation erben, werden betroffen sein; Töchter, die die Mutation erben, sind Überträgerinnen und werden meistens nicht ernsthaft betroffen sein.
Andere Familienmitglieder eines Probanden: Je nach Geschlecht, Verwandtschaftsbeziehung und Überträgerstatus der Eltern des Probanden können Onkel, Tanten und deren Kinder ein Risiko dafür haben, Überträger der Krankheit oder selbst davon betroffen zu sein.
Die Untersuchung von Familienmitgliedern mit einem solchen Risiko ist wichtig für die Betreuung und genetische Beratung, wird aber oft in ungenügender Weise durchgeführt. Mehrere Faktoren können hierfür verantwortlich sein:
- Die Feststellung der Diagnose bei einer betroffenen Person mit schwerer Behinderung kann für die Familie ein niederschmetterndes Ereignisse sein.
- Die molekulargenetische Analyse für klinische Zwecke steht noch nicht lange zur Verfügung und mag nicht überall bekannt sein.
- Krankenversicherungen können die Untersuchung eines Familienmitglieds mit Risiko ablehnen.
- Familienmitglieder mit einem Risiko lehnen die Untersuchung ab aus Furcht, bei positivem Ergebnis die Fähigkeit zu verlieren, eine Krankenversicherung zu bekommen oder zu behalten.
- Jemand weiß nicht genau, wer in der Familie ein Risiko haben könnte und scheut sich, bestimmte Familienmitglieder über das Risiko zu informieren.
Prävalenz der ALD
Die Prävalenz wird auf zwischen 1:20.000 und 1:50.000 geschätzt und scheint in allen ethnischen Gruppen ähnlich zu sein. In den Vereinigten Staaten wird die minimale Häufigkeit von identifizierten Hemizygoten auf 1:21.000 und diejenige von Hemizygoten und Heterozygoten zusammen auf 1:16.800 geschätzt.
Diagnose der ALD
Klinische Diagnose
Die Diagnose ALD sollte bei vier verschiedenen klinischen Konstellationen in Betracht gezogen werden:
- Knaben mit den Erscheinungen eines Aufmerksamkeitsdefizit-Syndroms (ADS), bei denen zusätzlich Zeichen bestehen von Demenz, zunehmenden Verhaltensstörungen, abnehmender Sehkraft, Schwierigkeiten beim Verständnis von Sprache, Verschlechterung der Handschrift, Koordinationsstörungen oder anderen neurologischen Auffälligkeiten.
- Junge oder mittelalte Männer mit zunehmenden Gangstörungen, Versteifungen oder Schwäche der Beine, Störungen der Schließmuskel- und der Sexualfunktion, mit oder ohne Nebennierenversagen oder Defiziten bei Intelligenz oder Verhalten.
- Alle männlichen Personen mit primärem Nebennierenrindenversagen, mit oder ohne Anzeichen neurologische Störungen.
- Mittelalte oder ältere Frauen mit fortschreitender Beinlähmung (Paraparese), Störungen der Schließmuskelkontrolle und Gefühlsstörungen hauptsächlich in den Beinen. Bei einer Frau ohne entsprechende Familienvorgeschichte kann die Diagnose einer ALD schwierig sein. Hier beruht die Diagnose auf klinischen Erscheinungen, am häufigsten einer fortschreitenden spastischen Paraparese in Verbindung mit verschiedenen Labortests.
Bildgebende Verfahren
MRT-Bilder des Gehirns sind beim männlichen Personen mit neurologischen Symptomen immer abnorm und stellen oft den ersten diagnostischen Hinweis dar. In etwa 85% der betroffenen Individuen zeigt das MRT ein charakteristisches Muster mit symmetrischer Verstärkung des T2-Signals in den parieio-okzipitalen Regionen des Gehirns und Kontrastverstärkung am fortschreitenden Rand der Läsionen.
Bei Personen, die hinsichtlich einer Mutation des ABCD1-Gens heterozygot sind, ist das MRT-Bild des Gehirns in weniger als 10% der Fälle abnorm.

Sehr Langkettige Fettsäuren (Very long-chain fatty acids, VLCFA)
Männliche Personen: wichtigster Labortests ist die Bestimmung der Konzentration der sehr langkettigen Fettsäuren (VLCFA) im Blutplasma. Die Spiegel der VLCFA sind bei 99,9% männlicher Personen mit X-ALD aller Altersstufen erhöht, ohne Rücksicht auf Vorhandensein oder Fehlen klinischer Symptome. Die drei untersuchten Testparameter sind: die Konzentration der Fettsäure C26:0, das Verhältnis C24:0/C22:0 und das Verhältnis C26:0/C22:0. Die Tab. 1 zeigt die mittleren Ergebnisse bei Normalpersonen, betroffenen männlichen Personen und weiblichen Überträgern [Valianpour et al., 2003].
Die Bestimmung der VLCFA ist sehr anspruchsvoll und wird daher auf der ganzen Welt nur in wenigen Laboratorien durchgeführt.
| Normalpersonen | Männliche Personen mit X-ALD | Überträgerinnen | |
| C26:0 µmol/L | 0.67 +/- 0.13 | 2.94 +/- 0.87 | 1.54 +/- 0.72 |
| C24:0/C22:0 ratio | 0.86 +/- 0.13 | 1.52 +/- 0.21 | 1.18 +/- 0.15 |
| C26:0/C22:0 ratio | 0.01 +/- 0.003 | 0.05 +/- 0.02 | 0.02 +/- 0.01 |
| Tabell 1: Konzentrationen der VLCFA im Blut von Normalpersonen und X-ALD-Patienten, Bestimmung mittels Electrospray-Ionisationsmassenspektrometrie (ESI-MS) (Valianpour et al., 2003). | |||

Wichtiger Kommentar von Dr. Ann Moser: Lorenzos Öl, eine Mischung von Erucasäure und Ölsäure, wird therapeutisch verwendet um die Spiegel der VLCFA in Blut zu normalisieren. Bei der Messung der VLCFA im Blutplasma berichtet das Labor für peroxisomale Krankheiten am Kennedy Krieger Institute in Baltimore routinemäßig auch über die Spiegel der Erucasäure (C22:1). Gewisse beim Kochen verwendete Öle, z.B. Senfsaatöl, haben natürlicherweise einen hohen Gehalt von Erucasäure und können im Blut zu einer Erhöhung führen, die ähnlich ist wie diejenige unter einer Therapie mit Lorenzos Öl.
Weibliche Personen: Erhöhte Konzentrationen von VLCFA im Plasma und/oder kultivierten Hautfibroblasten finden sich in etwa 85% weiblicher Personen; 15% nachgewiesener Überträgerinnen haben normale Plasmaspiegel der VLCFA. Durchschnittliche Ergebnisse der Bestimmung der VLCFA bei obligaten Heterozygoten enthält die Tabelle 1. Mit einer veröffentlichten Diskriminanzfunktion Moser AB et al (1999) ist es nicht möglich, alle Überträgerinnen vom Bereich der Kontrollpersonen abzugrenzen, siehe Abbildung. Frauen sollten genetisch getestet werden, wenn eine ALD vermutet wird und die Spiegel der VLCFA normal sind.
Mutationsanalyse (Molekulargenetik)
Das ABCD1-Gen ist das einzige Gen von Bedeutung bei der ALD. Mehr als 500 verschiedene Mutationen sind im ABCD1-Gen identifiziert worden [Kemp et al 2001]. Die meisten ALD-Stammbäume haben eine jeweils einzigartige Mutation. Ein Katalog der bekannten ABCD1-Gene findet sich auf dieser Website.
Boehm und Mitarbeiter haben einen zuverlässigen diagnostischen DNA-Test für ALD entwickelt und validiert auf der Basis einer „non-nested genomic amplification“, gefolgt von einer Sequenzanalyse unter Verwendung von Fluoreszenzfarben-Primern. Die Methode deckt alle kodierendem Exone und benachbarten Intron-Exon-Abschnitte in 10 getrennten Ampliconen ab [Boehm et al., 1999]. Diese Arbeitsvorschrift erlaubt die hoch zuverlässige Bestimmung des Überträgerstatus von Frauen mit einem Risiko, die ALD weiterzugeben und ist in einem klinisch-diagnostischen Labor anwendbar. Diese Methode ist inzwischen die sequenzbasierte Analyse der Wahl in vielen Labors weltweit.

Identifizierung von Überträgerinnen
Die Untersuchung einer weiblichen Person mit dem Risiko für den Überträgerstatus verläuft in zwei Schritten. Zuerst werden die sehr langkettigen Fettsäuren (VLCFA) bestimmt; wenn sie erhöht sind, handelt es sich um eine Überträgerin. Da aber 15% der Überträgerinnen normale VLCFA-Plasmaspiegel der haben, muss bei weiblichen Personen mit normalen VLCFA-Plasmaspiegeln aus Familien, in denen eine krankheitsverursachende Mutation des ABCD1-Gens identifiziert wurde, eine molekulargenetische Analyse durchgeführt werden. In einem Labor konnten bei 97% von obligaten Überträgerinnen die Mutationen vollständig identifiziert werden (n=29).
Erweiterte Familienuntersuchungen
Je nach Geschlecht, Verwandtschaftsbeziehung und Überträgerstatus der Eltern des Probanden können Onkel, Tanten und deren Kinder ein Risiko dafür haben, Überträger der Krankheit oder selbst davon betroffen zu sein.
Die Untersuchung von Familienmitgliedern mit einem solchen Risiko ist wichtig für die Betreuung und genetische Beratung, wird aber oft in ungenügender Weise durchgeführt. Mehrere Faktoren können hierfür verantwortlich sein:
- Die Feststellung der Diagnose bei einer betroffenen Person mit schwerer Behinderung kann für die Familie ein niederschmetterndes Ereignisse sein.
- Die molekulargenetische Analyse für klinische Zwecke steht noch nicht lange zur Verfügung und mag nicht überall bekannt sein.
- Krankenversicherungen können die Untersuchung eines Familienmitglieds mit Risiko ablehnen.
- Familienmitglieder mit einem Risiko lehnen die Untersuchung ab aus Furcht, bei positivem Ergebnis die Fähigkeit zu verlieren, eine Krankenversicherung zu bekommen oder zu behalten.
- Jemand weiß nicht genau, wer in der Familie ein Risiko haben könnte und scheut sich, bestimmte Familienmitglieder über das Risiko zu informieren.
Pränatale Untersuchung
Eine pränatale Untersuchung ist möglich bei Schwangerschaften von Überträgerinnen, bei denen das Risiko für einen von der Krankheit betroffenen Knaben 25% beträgt (oder 50%, wenn das männliche Geschlecht des Fetus bekannt ist). Üblicherweise wird zuerst das Geschlecht bestimmt durch Karyotypisierung fetaler Zellen, die aus Chorionzotten in etwa der 10.-12. Schwangerschaftswoche gewonnen werden (die Schwangerschaftsdauer bestimmt sich in Wochen nach dem ersten Tag der letzten Regelblutung oder durch Ultraschallmessungen). Fetale Zellen können auch in der 16.-18. Schwangerschaftswoche mittels Amniozentese aus Fruchtwasser gewonnen werden. Wenn der Karyotyp 46,XY (männlich) ist und die krankheitsverursachende Mutation bei einem Familienmitglied identifiziert wurde, kann DNA aus fetale Zellen auf die bekannte krankheitsverursachende Mutation untersucht werden.
Wenn die Mutationsanalyse nicht möglich ist, können sehr langkettigen Fettsäuren (VLCFA) in gezüchteten Amniozyten oder gezüchteten Zellen aus Chorionzotten (chorionic villus cells, CVC) gemessen werden [Wanders et al 1998, Moser AB et al 1999]. (Bei diesem Verfahren wurde über falsch negative Ergebnisse berichtet, die möglicherweise auf technischen Schwierigkeiten beruhten.)
Proteinanalyse
Das Gen-Produkt des ABCD1-Gens, ALD-Protein oder ALDP, ist mit der Methode der Immunfluoreszenzanalyse bei etwa 70% betroffener Individuen nicht nachweisbar. Aus nicht gut verstandenen Gründen kann das Gen-Produkt sogar bei Individuen mit Missense-Mutationen nicht nachweisbar sein. Wichtigste biochemische Abnormität ist die Akkumulation gesättigter sehr langkettiger Fettsäuren, besonders von Hexakosansäure (C26:0) und Tetrakosansäure (C24:0), als Folge einer verminderten Fähigkeit diese Stoffe abzubauen, was normalerweise in den Peroxisomen stattfindet. ALDP (ALD-Protein) transportiert die VLCFA aus dem Zytosol ins Peroxisom.

Nachweis von ALDP mittels Immunofluoreszenz: in Fibroblasten einer Kontrollperson (links) zeigt die helle punktförmige Färbung die Anwesenheit von ALDP in Peroxisomen an. In der Mitte Fibroblasten eines männlichen Patienten mit ALD und einer Mutation, die die Stabilität von ALDP vermindert. Hier ist keine punktförmige Färbung sichtbar. Rechts Fibroblasten einer ALD-Überträgerin aus derselben Familie wie der männliche Patient. Das ABCD1-Gen liegt auf dem X-Chromosom. Weibliche Personen haben zwei X-Chromosomen, doch ist in jeder Zelle nur eines der X-Chromosomen aktiv. Die Zellen mit punktförmiger Färbung sind solche mit einer aktiven Kopie des normalen ABCD1-Gens, während diejenigen ohne punktförmige Färbung eine aktive Kopie des ABCD1-Gens mit Mutation besitzen.
Bilder von Dr. Merel Ebberink
Klinische Bilder
Die geschlechtsgebundene Adrenoleukodystrophie (ALD) ist eine Krankheit, die in vielen verschiedenen Formen (Phänotypen) zutage treten kann; sieben verschiedene Phänotypen wurde bei männlichen Patienten und fünf bei weiblichen Patienten beschrieben. Alle diese Phänotypen werden durch genetische Fehler (Mutationen) im ABCD1-Gen verursacht. Innerhalb einer Familie haben alle ALD-Patienten dieselbe Mutation. Bisher wurden über 500 verschiedene Mutationen gefunden – die meisten davon sind in dieser Datenbasis gesammelt. Unglücklicherweise haben die Mutationen keinen Voraussagewert im Hinblick auf den klinischen Verlauf bei einem bisher asymptomatischen Patienten. Bis heute wurden keine Marker mit einem solchen Voraussagewert identifiziert; weder die Blutspiegel der sehr langkettigen Fettsäuren (VLCFA), noch die Mutationen im Gen, noch die Familienvorgeschichte im Hinblick auf ALD-Fälle, noch das Vorhandensein oder Fehlen von ALD-Protein in Zellkulturen.
Prävalenz (Häufigkeit) von ALD: eine große Studie aus dem Kennedy Krieger Institute und der Mayo Clinic führte zu dem Schluss, dass (wenigstens in den vereinigten Staaten) ALD etwa einmal unter 21.000 männlichen Neugeborenen auftritt und der Überträgerzustand für ALD etwa einmal unter 14.000 weiblichen Neugeborenen. Diese Angaben stimmen mit Häufigkeiten überein, die zuvor aus Frankreich berichtet worden waren. Die Häufigkeit von ALD über alles ist etwa eins auf 17.000 Lebendgeborene. ALD wurde in allen europäischen und lateinamerikanischen Ländern, China, Japan, Indien, Israel und Saudi-Arabien identifiziert, und auch in allen ethnischen Gruppen, eingeschlossen Afro-Amerikaner, eingeborene Amerikaner und Maori.
Frauen mit ALD: Obwohl es eine hohe Zahl von weiblichen Personen mit ALD gibt (Überträgerinnen), werden viele davon nicht diagnostiziert, weil die meisten Überträgerinnen bis ins mittlere Lebensalter symptomlos bleiben. Dazu kommt, dass viele Frauen mit Symptomen nicht diagnostiziert werden, wenn sie keinen betroffenen männlichen Verwandten mit Symptomen haben. Kürzlich hat eine Studie aus dem Kennedy Krieger Institute gezeigt, dass von 616 identifizierten Familien mit ALD nur in 16 Fällen der erste in der Familie identifizierte Patient weiblich war. Die große Mehrheit der Überträgerinnen wird erst identifiziert, nachdem ein männlicher Patient in der Familie diagnostiziert wurde (im Rahmen von ausgedehntem Suchprogrammen in Familien). Die beste Methode, um bei Frauen mit Verdacht auf X-ALD den Überträgerzustand festzustellen, ist die genetische Analyse des ALD-verursachenden Gens. Aber um dieses Verfahren wirklich verlässlich zu machen, muss die Mutation in der betreffenden Familie bei einem betroffenen männlichen Verwandten oder bei einer obligaten Überträgerin festgestellt worden sein. Eine obligate Überträgerin ist eine Frau, die entweder einen Vater mit diagnostizierter ALD hat oder die zwei Kinder hat, bei denen die ALD genetisch bewiesen ist.
Wir empfehlen, dass Frauen mit einem Risiko für den Überträgerstatus über die Möglichkeit der genetischen Tests aufgeklärt werden, wenn sie das Fortpflanzungsalter erreichen. Sie können dann informiert eine Entscheidung treffen, ob sie getestet werden möchten, und man kann ihnen dann auch die Möglichkeit geben, mit einer genetischen Beratungsstelle in Kontakt zu treten.
Klinische Beschreibung weiblicher Personen
Asymptomatische ALD: Das Vorliegen von ALD wurde hier durch eines der folgenden Kriterien gezeigt: Nachweis erhöhter Spiegel von VLCFA, einer im ABCD1-Gen identifizierten Mutation, oder sie hat einen Vater mit ALD oder mindestens zwei Kinder mit ALD. Danebenbestehen keine Anzeichen einer Störung der Nebennieren oder des Nervensystems. Die Anzahl asymptomatischer Überträgerinnen nimmt mit dem Alter ab. Die Mehrheit von weniger als 30 Jahre alten Frauen haben keine neurologischen Symptome.
Milde Myelopathie: Leicht herabgesetzter Vibrationssinn und etwas gesteigerte Reflexe an den Beinen. Dies beruht auf leichteren Schäden am Myelin (der Isolation um die Nervenfasern). Dieser Myelinschaden wirkt sich auf die Erregungsleitung der Axone der Nerven aus (der langen Nervenfasern, die Signale vom Rückenmark zu den Muskeln und anderen Körperteilen transportieren). Dies ist etwa in 50% der Überträgerinnen im Alter über 40 Jahre der Fall.
Mäßige bis schwere Myeloneuropathie: Die Symptome sind ähnlich wie die bei männlichen Patienten mit Adrenomyeloneuropathie (AMN, siehe unten), aber milder. Diese Form findet sich in etwa 15% der Überträgerinnen über 40 Jahre.
Zerebrale Beteiligung: Diese Form gleich der kindlichen zerebralen ALD (siehe unten). Sie ist bei Mädchen während der Kindheit sehr selten, es sind nur ein paar Einzelfälle bekannt. Bei Überträgerinnen im mittleren Alter oder darüber etwa bei 2%.
Klinisch in Erscheinung tretendes Nebennierenversagen: Addison-Krankheit (siehe unten). Sehr selten der Überträgerinnen jeden Alters, weniger als 1%.
Klinische Beschreibung männlicher Personen
Asymptomatisch oder präsymptomatisch: Einige Patienten, besonders solche, die durch Familien-Suchprogramme identifiziert wurden, haben unter Umständen niemals neurologische oder hormonelle Probleme. Bei ihnen wurde die ALD durch eines der folgenden Kriterien nachgewiesen: Das Vorliegen von ALD wurde bei ihnen durch eines der folgenden Kriterien gezeigt: Nachweis erhöhter Spiegel von VLCFA oder einer im ABCD1-Gen identifizierten Mutation. Kein Nachweis einer Störung der Nebennieren oder des Nervensystems. Die Anzahl asymptomatischer männlicher Personen nimmt mit dem Alter ab. Es ist wichtig, dass diese Patienten häufig von einem Arzt gesehen werden und die Nebennierenfunktion laufend untersucht wird. Für diese Patienten ist das Risiko neurologische Symptome zu entwickeln hoch. Diese Form ist häufig bei Jungen unter dem Alter von vier Jahren und sehr selten bei Männern über 40 Jahre.
Kindliche zerebrale ALD (childhood cerebral ALD, CCALD): Die Krankheit tritt am häufigsten im Alter zwischen vier und acht Jahren auf mit einem Maximum bei sieben Jahren. Diese Form kommt praktisch nie vor dem Alter von drei Jahren und sehr selten nach dem Alter von 15 Jahren vor. Betroffene Jungen zeigen ein abnormes Verhalten oder Lernschwierigkeiten, die häufig als Aufmerksamkeitsdefizit oder Hyperaktivitätsstörung diagnostiziert werden, die auf stimulierende Medikamente ansprechen. Diese Erscheinungen können Monate oder länger dauern, werden aber dann von Symptomen gefolgt, die eine zugrunde liegende ernstere Störung nahelegen.
Zu diesen Symptomen können Auffälligkeiten in der Schule gehören: Unaufmerksamkeit, Verschlechterung der Handschrift und Abfall der Schulleistungen; Schwierigkeiten beim Verstehen von Sprache (bei normaler Hörfähigkeit); Schwierigkeiten beim Lesen, bei der räumlichen Orientierung und beim Verständnis von Geschriebenem; Schwerfälligkeit; Sehstörungen und gelegentliches Doppeltsehen; aggressives und enthemmtes Verhalten. Die MRT-Untersuchung des Gehirns kann zu diesem Zeitpunkt eine erhebliche abnormen Befund ergeben, auch wenn die Symptome verhältnismäßig mild erscheinen. Bei einigen Jungen können Krämpfe das erste Krankheitszeichen sein. Die Geschwindigkeit des Fortschreitens der Krankheit ist unterschiedlich. Sie kann sehr rasch sein mit völliger Hilflosigkeit in sechs Monaten bis zwei Jahren, mit nachfolgendem Tod in unterschiedlichem Alter. Die meisten Patienten haben eine gestörte Nebennierenfunktion zu dem Zeitpunkt, an dem die neurologischen Störungen bemerkt werden. Ungefähr 35% der ALD-Patienten entwickelt die kindliche zerebrale ALD (CCALD).
Zerebrale ALD der Adoleszenz: Ähnlich wie CCALD. Der Beginn liegen zwischen dem Alter von 11 und 21 Jahren. Vorkommen bei 4-7% der Patienten.
Zerebrale ALD der Erwachsenen: Diese Form entwickelt sich in 2-5% der Patienten. Bei diesen Patienten wird oft fälschlich die Diagnose einer paranoiden Psychose, Schizophrenie oder einer anderen psychiatrischen Störung gestellt. Dieser Form geht keine AMN voraus (siehe unten). Es besteht eine entzündliche Reaktion der weißen Substanz (sichtbar im MRT). Das Voranschreiten der Krankheit verläuft demjenigen der kindlichen zerebralen Form parallel.
Adrenomyeloneuropathie (AMN): Typisches Erscheinungsbild ist ein Mann in den Zwanzigern oder in mittlerem Alter, bei dem sich eine fortschreitende Versteifung und Schwäche der Beine entwickelt, Störungen der Schließmuskelkontrolle und der Potenz. Alle Symptome schreiten über Jahrzehnte voran. Etwa 40-45% der Patienten mit AMN zeigen einen gewissen Grad von Gehirnbeteiligung im MRT oder bei klinischer Untersuchung. Bei 10-20% der Patienten mit AMN wird die Gehirnbeteiligung schwer progressiv und führt zu ernsten Störungen der Kognition und des Verhaltens, die in völliger Hilflosigkeit und Tod enden können. Etwa 70% der Männer mit Adrenomyeloneuropathie haben eine gestörte Nebennierenfunktion zum Zeitpunkt des Auftretens neurologischer Symptome. Etwa 45% dieser Patienten haben ein Risiko, zerebrale Beteiligung zu entwickeln.
Phänotyp “nur Addison” (Addison-only phenotype): Bei diesen männlichen Patienten treten Zeichen des Nebennierenversagens zwischen dem Alter von 2 Jahren und dem Erwachsenenalter auf, am häufigsten um das Alter von siebeneinhalb Jahren. Zu diesen Zeichen gehören unverständliches Erbrechen und Schwächeattacken oder Bewusstlosigkeit, die zur Diagnose der Addison-Krankheit führen. Bei einem Teil dieser Personen tritt aufgrund erhöhter Sekretion von ACTH eine verstärkte Pigmentation der Haut auf. Die meisten Personen, bei denen das Nebennierenversagen erstes Symptom ist, entwickeln Anzeichen einer AMN erst im mittleren Alter. Etwa 10% der ALD-Patienten haben diesen Phänotyp “nur Addison”.