
Publications Team ALD
Sex-specific newborn screening for X-Linked adrenoleukodystrophy.
Albersen M, van der Beek SL, Dijkstra IME, Alders M, Barendsen RW, Bliek J, Boelen A, Ebberink MS, Ferdinandusse S, Goorden SMI, Heijboer AC, Jansen M, Jaspers YRJ, Metgod I, Salomons GS, Vaz FM, Verschoof-Puite RK, Visser WF, Dekkers E, Engelen M, Kemp S.
J Inherit Metab Dis. 2022 Oct 18. doi: 10.1002/jimd.12571. Online ahead of print. PMID: 36256460
International Recommendations for the Diagnosis and Management of Patients With Adrenoleukodystrophy: A Consensus-Based Approach.
Engelen M, van Ballegoij WJC, Mallack EJ, Van Haren KP, Köhler W, Salsano E, van Trotsenburg ASP, Mochel F, Sevin C, Regelman MO, Tritos NA, Halper A, Lachmann RH, Davison J, Raymond GV, Lund T, Orchard PJ, Kuehl JS, Lindemans CA, Caruso P, Turk BR, Moser AB, Vaz FM, Ferdinandusse S, Kemp S, Fatemi A, Eichler FS, Huffnagel IC.
Neurology. 2022 Sep 29:10.1212/WNL.0000000000201374. doi: 10.1212/WNL.0000000000201374. Online ahead of print. PMID: 36175155
Treatment of cerebral adrenoleukodystrophy: allogeneic transplantation and lentiviral gene therapy.
Gupta AO, Raymond G, Pierpont EI, Kemp S, McIvor RS, Rayannavar A, Miller B, Lund TC, Orchard PJ.
Expert Opin Biol Ther. 2022 Sep;22(9):1151-1162. PMID: 36107226
Peroxisomal very long-chain fatty acid transport is targeted by herpesviruses and the antiviral host response.
Weinhofer I, Buda A, Kunze M, Palfi Z, Traunfellner M, Hesse S, Villoria-Gonzalez A, Hofmann J, Hametner S, Regelsberger G, Moser AB, Eichler F, Kemp S, Bauer J, Kühl JS, Forss-Petter S, Berger J.
Commun Biol. 2022 Sep 9;5(1):944. PMID: 36085307
Peroxisome Metabolism Contributes to PIEZO2-Mediated Mechanical Allodynia.
Gong Y, Laheji F, Berenson A, Qian A, Park SO, Kok R, Selig M, Hahn R, Sadjadi R, Kemp S, Eichler F.
Cells. 2022 Jun 4;11(11):1842. PMID: 35681537
Structure and Function of the ABCD1 Variant Database: 20 Years, 940 Pathogenic Variants, and 3400 Cases of Adrenoleukodystrophy.
Mallack EJ, Gao K, Engelen M, Kemp S.
Cells. 2022 Jan 14;11(2):283. PMID: 35053399
Biochemical Studies in Fibroblasts to Interpret Variants of Unknown Significance in the ABCD1 Gene.
van de Stadt SIW, Mooyer PAW, Dijkstra IME, Dekker CJM, Vats D, Vera M, Ruzhnikov MRZ, van Haren K, Tang N, Koop K, Willemsen MA, Hui J, Vaz FM, Ebberink MS, Engelen M, Kemp S, Ferdinandusse S.
Genes (Basel). 2021 Nov 30;12(12):1930. PMID: 34946879
Molecular Biomarkers for Adrenoleukodystrophy: An Unmet Need.
Honey MIJ, Jaspers YRJ, Engelen M, Kemp S, Huffnagel IC.
Cells. 2021 Dec 6;10(12):3427. PMID: 34943935
iBRET Screen of the ABCD1 Peroxisomal Network and Mutation-Induced Network Perturbations.
Lotz-Havla AS, Woidy M, Guder P, Friedel CC, Klingbeil JM, Bulau AM, Schultze A, Dahmen I, Noll-Puchta H, Kemp S, Erdmann R, Zimmer R, Muntau AC, Gersting SW.
J Proteome Res. 2021 Sep 3;20(9):4366-4380. PMID: 34383492
Endocrine dysfunction in adrenoleukodystrophy.
Engelen M, Kemp S, Eichler F.
Handb Clin Neurol. 2021;182:257-267. PMID: 34266597
The brain penetrant PPARγ agonist leriglitazone restores multiple altered pathways in models of X-linked adrenoleukodystrophy.
Rodríguez-Pascau L, Vilalta A, Cerrada M, Traver E, Forss-Petter S, Weinhofer I, Bauer J, Kemp S, Pina G, Pascual S, Meya U, Musolino PL, Berger J, Martinell M, Pizcueta P.
Sci Transl Med. 2021 Jun 2;13(596):eabc0555. PMID: 34078742
Metabolic rerouting via SCD1 induction impacts X-linked adrenoleukodystrophy.
Raas Q, van de Beek MC, Forss-Petter S, Dijkstra IM, Deschiffart A, Freshner BC, Stevenson TJ, Jaspers YR, Nagtzaam L, Wanders RJ, van Weeghel M, Engelen-Lee JY, Engelen M, Eichler F, Berger J, Bonkowsky JL, Kemp S.
J Clin Invest. 2021 Apr 15;131(8):e142500. PMID: 33690217
MRI surveillance of boys with X-linked adrenoleukodystrophy identified by newborn screening: Meta-analysis and consensus guidelines.
Mallack EJ, Turk BR, Yan H, Price C, Demetres M, Moser AB, Becker C, Hollandsworth K, Adang L, Vanderver A, Van Haren K, Ruzhnikov M, Kurtzberg J, Maegawa G, Orchard PJ, Lund TC, Raymond GV, Regelmann M, Orsini JJ, Seeger E, Kemp S, Eichler F, Fatemi A.
J Inherit Metab Dis. 2021 May;44(3):728-739. PMID: 33373467
Evolution of adrenoleukodystrophy model systems.
Montoro R, Heine VM, Kemp S, Engelen M.
J Inherit Metab Dis. 2021 May;44(3):544-553. PMID: 33373044
The variability conundrum in neurometabolic degenerative diseases.
van Karnebeek CDM, Richmond PA, van der Kloet F, Wasserman WW, Engelen M, Kemp S.
Mol Genet Metab. 2020 Dec;131(4):367-369. PMID: 33246824
Plasma NfL and GFAP as biomarkers of spinal cord degeneration in adrenoleukodystrophy.
van Ballegoij WJC, van de Stadt SIW, Huffnagel IC, Kemp S, Willemse EAJ, Teunissen CE, Engelen M.
Ann Clin Transl Neurol. 2020 Nov;7(11):2127-2136. PMID: 33047897
Targeting foam cell formation in inflammatory brain diseases by the histone modifier MS-275.
Zierfuss B, Weinhofer I, Buda A, Popitsch N, Hess L, Moos V, Hametner S, Kemp S, Köhler W, Forss-Petter S, Seiser C, Berger J.
Ann Clin Transl Neurol. 2020 Nov;7(11):2161-2177. PMID: 32997393
Comparison of the Diagnostic Performance of C26:0-Lysophosphatidylcholine and Very Long-Chain Fatty Acids Analysis for Peroxisomal Disorders.
Jaspers YRJ, Ferdinandusse S, Dijkstra IME, Barendsen RW, van Lenthe H, Kulik W, Engelen M, Goorden SMI, Vaz FM, Kemp S.
Front Cell Dev Biol. 2020 Jul 29;8:690. doi: 10.3389/fcell.2020.00690. PMID: 32903870
Postural Body Sway as Surrogate Outcome for Myelopathy in Adrenoleukodystrophy.
van Ballegoij WJC, van de Stadt SIW, Huffnagel IC, Kemp S, van der Knaap MS, Engelen M.
Front Physiol. 2020 Jul 17;11:786. PMID: 32765293
Multi-Omic Approach to Identify Phenotypic Modifiers Underlying Cerebral Demyelination in X-Linked Adrenoleukodystrophy.
Richmond PA, van der Kloet F, Vaz FM, Lin D, Uzozie A, Graham E, Kobor M, Mostafavi S, Moerland PD, Lange PF, van Kampen AHC, Wasserman WW, Engelen M, Kemp S, van Karnebeek CDM.
Front Cell Dev Biol. 2020 Jun 25;8:520. PMID: 32671069
Adrenoleukodystrophy Newborn Screening in the Netherlands (SCAN Study): The X-Factor.
Barendsen RW, Dijkstra IME, Visser WF, Alders M, Bliek J, Boelen A, Bouva MJ, van der Crabben SN, Elsinghorst E, van Gorp AGM, Heijboer AC, Jansen M, Jaspers YRJ, van Lenthe H, Metgod I, Mooij CF, van der Sluijs EHC, van Trotsenburg ASP, Verschoof-Puite RK, Vaz FM, Waterham HR, Wijburg FA, Engelen M, Dekkers E, Kemp S.
Front Cell Dev Biol. 2020 Jun 17;8:499. PMID: 32626714
Spinal cord atrophy as a measure of severity of myelopathy in adrenoleukodystrophy.
van de Stadt SIW, van Ballegoij WJC, Labounek R, Huffnagel IC, Kemp S, Nestrasil I, Engelen M.
J Inherit Metab Dis. 2020 Jul;43(4):852-860. PMID: 32077106
Longitudinal diffusion MRI as surrogate outcome measure for myelopathy in adrenoleukodystrophy.
Huffnagel IC, van Ballegoij WJC, Vos JMBW, Kemp S, Caan MWA, Engelen M.
Neurology. 2019 Dec 3;93(23):e2133-e2143. PMID: 31719133
Disease progression in women with X-linked adrenoleukodystrophy is slow.
Huffnagel IC, Dijkgraaf MGW, Janssens GE, van Weeghel M, van Geel BM, Poll-The BT, Kemp S, Engelen M.
Orphanet J Rare Dis. 2019 Feb 7;14(1):30. PMID: 30732635
Progression of myelopathy in males with adrenoleukodystrophy: towards clinical trial readiness.
Huffnagel IC, van Ballegoij WJC, van Geel BM, Vos JMBW, Kemp S, Engelen M.
Brain. 2019 Feb 1;142(2):334-343. PMID: 30535170
The Natural History of Adrenal Insufficiency in X-Linked Adrenoleukodystrophy: An International Collaboration.
Huffnagel IC, Laheji FK, Aziz-Bose R, Tritos NA, Marino R, Linthorst GE, Kemp S, Engelen M, Eichler F.
J Clin Endocrinol Metab. 2019 Jan 1;104(1):118-126. PMID: 30252065
Comparison of C26:0-carnitine and C26:0-lysophosphatidylcholine as diagnostic markers in dried blood spots from newborns and patients with adrenoleukodystrophy.
Huffnagel IC, van de Beek MC, Showers AL, Orsini JJ, Klouwer FCC, Dijkstra IME, Schielen PC, van Lenthe H, Wanders RJA, Vaz FM, Morrissey MA, Engelen M, Kemp S.
Mol Genet Metab. 2017 Dec;122(4):209-215. PMID: 29089175
Lipid-induced endoplasmic reticulum stress in X-linked adrenoleukodystrophy.
van de Beek MC, Ofman R, Dijkstra I, Wijburg F, Engelen M, Wanders R, Kemp S.
Biochim Biophys Acta Mol Basis Dis. 2017 Sep;1863(9):2255-2265. PMID: 28666219
Adrenoleukodystrophy – neuroendocrine pathogenesis and redefinition of natural history.
Kemp S, Huffnagel IC, Linthorst GE, Wanders RJ, Engelen M.
Nat Rev Endocrinol. 2016 Oct;12(10):606-15. PMID: 27312864
26:0-Carnitine Is a New Biomarker for X-Linked Adrenoleukodystrophy in Mice and Man.
van de Beek MC, Dijkstra IM, van Lenthe H, Ofman R, Goldhaber-Pasillas D, Schauer N, Schackmann M, Engelen-Lee JY, Vaz FM, Kulik W, Wanders RJ, Engelen M, Kemp S.
PLoS One. 2016 Apr 28;11(4):e0154597. PMID: 27124591
Pathogenicity of novel ABCD1 variants: The need for biochemical testing in the era of advanced genetics.
Schackmann MJ, Ofman R, van Geel BM, Dijkstra IM, van Engelen K, Wanders RJ, Engelen M, Kemp S.
Mol Genet Metab. 2016 Jun;118(2):123-7. PMID: 27067449
Hematopoietic cell transplantation does not prevent myelopathy in X-linked adrenoleukodystrophy: a retrospective study.
van Geel BM, Poll-The BT, Verrips A, Boelens JJ, Kemp S, Engelen M.
J Inherit Metab Dis. 2015 Mar;38(2):359-61. PMID: 25488625
X-linked adrenoleukodystrophy: pathogenesis and treatment.
Engelen M, Kemp S, Poll-The BT.
Curr Neurol Neurosci Rep. 2014 Oct;14(10):486. PMID: 25115486
Reply: Age-dependent penetrance among females with X-linked adrenoleukodystrophy.
Engelen M, Barbier M, Dijkstra IM, Schür R, de Bie RM, Verhamme C, Dijkgraaf MG, Aubourg PA, Wanders RJ, van Geel BM, de Visser M, Poll-The BT, Kemp S.
Brain. 2015 Feb;138(Pt 2):e326. PMID: 25149411
X-linked adrenoleukodystrophy in women: a cross-sectional cohort study.
Engelen M, Barbier M, Dijkstra IM, Schür R, de Bie RM, Verhamme C, Dijkgraaf MG, Aubourg PA, Wanders RJ, van Geel BM, de Visser M, Poll-The BT, Kemp S.
Brain. 2014 Mar;137(Pt 3):693-706. PMID: 24480483
Comment on the paper “Effect of statin treatment on adrenomyeloneuropathy with cerebral inflammation: a revisit”.
Engelen M, Ofman R, Dijkgraaf M, van Geel B, de Visser M, Wanders R, Poll-The BT, Kemp S.
Clin Neurol Neurosurg. 2013 Nov;115(11):2401-2. PMID: 24018110
X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management.
Engelen M, Kemp S, de Visser M, van Geel BM, Wanders RJ, Aubourg P, Poll-The BT.
Orphanet J Rare Dis. 2012 Aug 13;7:51. PMID: 22889154
Bezafibrate lowers very long-chain fatty acids in X-linked adrenoleukodystrophy fibroblasts by inhibiting fatty acid elongation.
Engelen M, Schackmann MJ, Ofman R, Sanders RJ, Dijkstra IM, Houten SM, Fourcade S, Pujol A, Poll-The BT, Wanders RJ, Kemp S.
J Inherit Metab Dis. 2012 Nov;35(6):1137-45. PMID: 22447153
Bezafibrate for X-linked adrenoleukodystrophy.
Engelen M, Tran L, Ofman R, Brennecke J, Moser AB, Dijkstra IM, Wanders RJ, Poll-The BT, Kemp S.
PLoS One. 2012;7(7):e41013. PMID: 22911730
X-linked adrenomyeloneuropathy due to a novel missense mutation in the ABCD1 start codon presenting as demyelinating neuropathy.
Engelen M, van der Kooi AJ, Kemp S, Wanders RJ, Sistermans EA, Waterham HR, Koelman JT, van Geel BM, de Visser M.
J Peripher Nerv Syst. 2011 Dec;16(4):353-5. PMID: 22176151
Lovastatin in X-linked adrenoleukodystrophy.
Engelen M, Ofman R, Dijkgraaf MG, Hijzen M, van der Wardt LA, van Geel BM, de Visser M, Wanders RJ, Poll-The BT, Kemp S.
N Engl J Med. 2010 Jan 21;362(3):276-7. PMID: 20089986
Cholesterol-deprivation increases mono-unsaturated very long-chain fatty acids in skin fibroblasts from patients with X-linked adrenoleukodystrophy.
Engelen M, Ofman R, Mooijer PA, Poll-The BT, Wanders RJ, Kemp S.
Biochim Biophys Acta. 2008 Mar;1781(3):105-11. PMID: 18206987
Characterization of the human omega-oxidation pathway for omega-hydroxy-very-long-chain fatty acids.
Sanders RJ, Ofman R, Dacremont G, Wanders RJ, Kemp S.
FASEB J. 2008 Jun;22(6):2064-71. PMID: 18182499
X-linked adrenoleukodystrophy: very long-chain fatty acid metabolism, ABC half-transporters and the complicated route to treatment.
Kemp S, Wanders RJ.
Mol Genet Metab. 2007 Mar;90(3):268-76. doi: 10.1016/j.ymgme.2006.10.001. Epub 2006 Nov 7.
PMID: 17092750
Omega-oxidation of very long-chain fatty acids in human liver microsomes. Implications for X-linked adrenoleukodystrophy.
Sanders RJ, Ofman R, Duran M, Kemp S, Wanders RJA.
J Biol Chem. 2006 May 12;281(19):13180-13187. doi: 10.1074/jbc.M513481200. Epub 2006 Mar 17.
PMID: 16547005
Gene redundancy and pharmacological gene therapy: implications for X-linked adrenoleukodystrophy.
Kemp S, Wei HM, Lu JF, Braiterman LT, McGuinness MC, Moser AB, Watkins PA, Smith KD.
Nat Med. 1998 Nov;4(11):1261-8. doi: 10.1038/3242.
PMID: 9809549
Editorial board of ALD info
 Marc Engelen received his M.D. from the University of Amsterdam in the Netherlands in 2002. He was trained as a neurologist and subsequently subspecialized in pediatric neurology at the Amsterdam University Medical Center. In 2012, Dr. Engelen earned his Ph.D. on “Translational studies in adrenoleukodystrophy”. He is currently the head of the Department of Pediatrics at the Amsterdam UMC. Marc has a special interest in peroxisomal diseases. In 2015, the Amsterdam UMC was designated as the national expert center these diseases. That same year, he started the prospective ALD patient cohort, the “Dutch ALD cohort,” which now contains natural history data on more than 180 ALD patients with more than 8 years of follow-up. This has provided insight into the rate of disease progression in ALD in men and women, and a new insights into the occurrence of specific symptoms. These data have significantly contributed to clinical trial readiness and are being used for clinical trial design and prospective biomarker research.
Marc Engelen received his M.D. from the University of Amsterdam in the Netherlands in 2002. He was trained as a neurologist and subsequently subspecialized in pediatric neurology at the Amsterdam University Medical Center. In 2012, Dr. Engelen earned his Ph.D. on “Translational studies in adrenoleukodystrophy”. He is currently the head of the Department of Pediatrics at the Amsterdam UMC. Marc has a special interest in peroxisomal diseases. In 2015, the Amsterdam UMC was designated as the national expert center these diseases. That same year, he started the prospective ALD patient cohort, the “Dutch ALD cohort,” which now contains natural history data on more than 180 ALD patients with more than 8 years of follow-up. This has provided insight into the rate of disease progression in ALD in men and women, and a new insights into the occurrence of specific symptoms. These data have significantly contributed to clinical trial readiness and are being used for clinical trial design and prospective biomarker research.

Julie Cohen is a certified genetic counselor with over 15 years of experience in clinical practice and research. She specializes in neurogenetics and leukodystrophies. She is the Director of Genetic Counseling Services at the Kennedy Krieger Institute and an Associate Professor of Clinical Neurology and Genetic Medicine at the Johns Hopkins University School of Medicine. Julie is the dedicated genetic counselor for the Moser Center for Leukodystrophies at Kennedy Krieger, where she provides comprehensive care to individuals and families affected by ALD and other leukodystrophies. She has particular expertise in the genetics of ALD, including interpreting ABCD1 variants and their clinical implications. Julie is a member of the Genomics Core of the Global Leukodystrophy Initiative Clinical Trials Network (GLIA-CTN) and a founding member of the ClinGen Leukodystrophy and Leukoencephalopathy Genomic Expert Curation Panel. Julie earned her graduate degree in genetic counseling from the Johns Hopkins Bloomberg School of Public Health and completed her training at the National Human Genome Research Institute at the NIH in 2009.
 Rachel Salzman is the Chief Scientific Officer of the Stop ALD Foundation (SALD), a position she has held since 2001. SALD is a nonprofit medical research organization dedicated to using entrepreneurial approaches and innovative methods to find effective therapies, cures and prevention for adrenoleukodystrophy. The Foundation’s biomedical interests include gene therapy, hematopoietic stem cells, mesenchymal and other adult stem cells, genomics, and small molecules. Rachel also consults with large pharmaceutical and biotechnology companies in the area of drug development, including preclinical and clinical analysis in a variety of therapeutic areas. This consulting includes advising animal health developers and the food manufacturing industry on nutritional supplements, preventive healthcare, and animal housing conditions welfare practices. Prior to these roles, Rachel worked in private veterinary practice for over 10 years in both large and small animal medicine. She holds a D.V.M. from Oklahoma State University and a B.S. from Rutgers University.
Rachel Salzman is the Chief Scientific Officer of the Stop ALD Foundation (SALD), a position she has held since 2001. SALD is a nonprofit medical research organization dedicated to using entrepreneurial approaches and innovative methods to find effective therapies, cures and prevention for adrenoleukodystrophy. The Foundation’s biomedical interests include gene therapy, hematopoietic stem cells, mesenchymal and other adult stem cells, genomics, and small molecules. Rachel also consults with large pharmaceutical and biotechnology companies in the area of drug development, including preclinical and clinical analysis in a variety of therapeutic areas. This consulting includes advising animal health developers and the food manufacturing industry on nutritional supplements, preventive healthcare, and animal housing conditions welfare practices. Prior to these roles, Rachel worked in private veterinary practice for over 10 years in both large and small animal medicine. She holds a D.V.M. from Oklahoma State University and a B.S. from Rutgers University.
 Virginie Bonnamain is currently a Clinical Scientist at Glycostem Therapeutics, where she leads clinical and project management activities for NK cell-based immunotherapy trials in oncology indications. Virginie has more than 12 years of experience in R&D and project management, in the life sciences, including 6 years in drug development, rare diseases and cell & gene therapy, acquired in various academic institutions (University of Nantes Medical School, Mount Sinai School of Medicine and the Cleveland Clinic) and biotech companies. In particular, she worked as Clinical Project Manager for a gene therapy trial for adrenoleukodystrophy and also contributed to the development of a European academic consortium for adrenomyeloneuropathy, aimed at improving patient care and understanding of the disease. Virginie received an MSc in Biological & Medical Sciences in 2006 and a PhD in Neuroimmunology in 2009 from the University of Nantes, France. She has published 18 scientific publications/book chapters.
Virginie Bonnamain is currently a Clinical Scientist at Glycostem Therapeutics, where she leads clinical and project management activities for NK cell-based immunotherapy trials in oncology indications. Virginie has more than 12 years of experience in R&D and project management, in the life sciences, including 6 years in drug development, rare diseases and cell & gene therapy, acquired in various academic institutions (University of Nantes Medical School, Mount Sinai School of Medicine and the Cleveland Clinic) and biotech companies. In particular, she worked as Clinical Project Manager for a gene therapy trial for adrenoleukodystrophy and also contributed to the development of a European academic consortium for adrenomyeloneuropathy, aimed at improving patient care and understanding of the disease. Virginie received an MSc in Biological & Medical Sciences in 2006 and a PhD in Neuroimmunology in 2009 from the University of Nantes, France. She has published 18 scientific publications/book chapters.
 Stephan Kemp is a Full Professor at the Amsterdam University Medical Center, University of Amsterdam, The Netherlands. He was trained as a translational researcher at the Johns Hopkins University/Kennedy Krieger Institute, Baltimore, Maryland, USA and received his Ph.D. from the University of Amsterdam in 1999. Stephan has more than 25 years of experience in adrenoleukodystrophy research and has published more than 100 papers and book chapters on adrenoleukodystrophy. In 1999, together with Dr. Hugo Moser, he initiated the ABCD1 variant registry (www.x-ald.nl), which moved to www.adrenoleukodystrophy.info in 2017. He is actively involved in patient organizations, serving on medical and scientific boards in Europe and the USA, and as a board member of ALD Connect (USA). Dr. Kemp led the development of a boys-only screening algorithm for ALD as part of the Dutch newborn screening program, leading to nationwide screening from October 1, 2023. He is the recipient of the 2015 AMC Societal Impact Award. His research interests are inherited neurometabolic diseases and newborn screening.
Stephan Kemp is a Full Professor at the Amsterdam University Medical Center, University of Amsterdam, The Netherlands. He was trained as a translational researcher at the Johns Hopkins University/Kennedy Krieger Institute, Baltimore, Maryland, USA and received his Ph.D. from the University of Amsterdam in 1999. Stephan has more than 25 years of experience in adrenoleukodystrophy research and has published more than 100 papers and book chapters on adrenoleukodystrophy. In 1999, together with Dr. Hugo Moser, he initiated the ABCD1 variant registry (www.x-ald.nl), which moved to www.adrenoleukodystrophy.info in 2017. He is actively involved in patient organizations, serving on medical and scientific boards in Europe and the USA, and as a board member of ALD Connect (USA). Dr. Kemp led the development of a boys-only screening algorithm for ALD as part of the Dutch newborn screening program, leading to nationwide screening from October 1, 2023. He is the recipient of the 2015 AMC Societal Impact Award. His research interests are inherited neurometabolic diseases and newborn screening.
Sitemap of the ALD website
The main language of adrenoleukodystrophy.info is English. But from an increasing number of pages translations in Español, Deutch, Français, or Nederlands are available.
English
Variants & Biochemistry
Variants in ABCD1
Large deletions
Variants statistics
Pseudogenes & Genetic Testing
Pathogenic variants & ALD Protein Stability
ALDP orthologs
ABCD proteins
Very long-chain fatty acids
Origin and Metabolism of VLCFA
Biochemical defect
ABCD1 coding region
The ABCD1 gene
Clinical & Diagnosis
Facts about ALD
Newborn screening
Recommendations for management
Diagnosis of ALD
Women with ALD
Clinical presentations
History of ALD
Treatment options
Hematopoietic stem cell transplantation
Gene Therapy for ALD
Lorenzo’s oil
Lovastatin in ALD
Adrenal insufficiency
The adrenal gland
Lorenzo Odone
Español
Ácidos Grasos de Cadena Muy Larga
Datos sobre la ALD
Cribado neonatal
Guía para el asesoramiento
Diagnóstico de ALD
Historia de ALD
Mujeres con ALD
Presentaciones clínicas
Trasplante de células madre hematopoyéticas
Terapia génica para la ALD
Aceite de Lorenzo
Lovastatina en ALD
Insuficiencia suprarrenal
La glándula suprarrenal
Lorenzo Odone
Deutch
Sehr langkettige Fettsäuren
Tatsachen zur ALD
Neugeborenenscreening
Genetik und genetische Beratung
Diagnose der ALD
Klinische Bilder
Français
Acides Gras à Très Longue Chaîne
Les faits sur l’ALD
Dépistage néonatal
Présentations cliniques
Les femmes ayant une ALD
Histoire de l’ALD
Thérapie Génique pour l’ALD
L’huile de Lorenzo
La lovastatine pour l’ALD
L’insuffisance surrénale
Lorenzo Odone
Nederlands
Zeer langketen vetzuren
Feiten over ALD
Hielprik screening
Vrouwen met ALD
Lovastatine en ALD
Gentherapie voor ALD
Behandeling van ALD
General
Hugo Moser
Variants of Uncertain Significance
Editorial board
Sitemap
Acknowledgements
Disclaimer
Contact
Acknowledgements
ALD info is a community driven project. Its strength lies in the sharing of information and the collaboration with many ALD experts with different fields of expertise.
We are very thankful to the following persons:
Website
John Hirschbeck Memorial Fund (for funding the first version of the website).
Marcel Mattijssen (for funding the hosting of the website (1999 – 2003)).
Netherlands ALD Patient Organization (for funding several years of the hosting cost (2003 – 2012)).
Ted van Geest (GoedGedaan) (for sponsoring the hosting of the website (since 2012) and his continuous help with the website and all the other stuff that keeps a website functional & running).
Scientific contributions
Johanna Assies, M.D., Ph.D.
Wouter van Ballegoij, M.D.
Johannes Berger, Ph.D.
Virginie Bonnamain, Ph.D.
Marc Engelen, M.D., Ph.D.
Gabor Linthorst, M.D.
Ann Moser, B.A.
Hugo Moser, M.D.
Charles Peters, M.D.
Gerald Raymond, M.D.
Rachel Salzman, D.V.M. Rachel Salzman, DVM (CSO, The Stop ALD Foundation)
Steven J Steinberg, Ph.D.
Björn van Geel, M.D., Ph.D.
Paul Watkins, M.D., Ph.D.
for their scientific contributions.
Translations to French, German and Spanish
Nerea Montedeoca Vázquez (Biomedicine student)
Cyntia Amorosi, Ph.D.
Alfried Kohlschütter M.D.
Elise Saunier Vivar, Ph.D. (European Leukodystrophy Foundation, ELA)
Carmen Sever (ELA España)
Disclaimer
The purpose of the adrenoleukodystrophy website is to provide general educational information about ALD. Our goal is to cover as many aspects of ALD as possible. This database and the information contained herein is not intended to be a substitute for professional medical advice, diagnosis or treatment. Please do not use our website for diagnosis or treatment of ALD. While we provide information, always consult your professional healthcare provider for specific disease-related questions or concerns.
Contributors are responsible for the reliability of the unverified data published on this website. Although considerable effort has been made to ensure the high quality of the ABCD1 Variant Database, the laboratories/investigators identifying these variants make no warranties, express or implied, regarding the accuracy of the information or its suitability for any particular purpose. Users should exercise caution for several reasons:
- There is no phenotype/genotype correlation in ALD. Prediction of disease course based on a genetic variant is not possible, even within individual families.
- The ‘variant’ may be a technical artifact.
- Many variants of uncertain significance (VUS) lack experimental evidence that the variant is not pathogenic – or benign.
- Due to the high percentage of unique variants in the ABCD1 gene, the pathogenicity of many variants has not been confirmed by independent evidence.
- Results from the Grey Zone Project are intended for research and diagnostic orientation purposes only and should always be discussed with a physician experienced in ALD or inherited metabolic disorders.
Unpublished variants may not be used for publication purposes without prior permission from the database editor and the laboratories/investigators who identified and reported these variants.
This website uses the web analytics tool Matomo for website statistics.
Contact
For comments, questions, or more information, please contact:
Stephan Kemp, Ph.D.
Full Professor of Inherited Neurometabolic Diseases and Newborn Screening
Department of Laboratory Medicine
Laboratory Genetic Metabolic Diseases
Amsterdam UMC – University of Amsterdam
The Netherlands
s.kemp@amsterdamumc.nl
Learn more about us and our research at: Team ALD

Facts about ALD
Authors: Marc Engelen, M.D., Ph.D., Rachel Salzman, D.V.M. (CSO, The Stop ALD Foundation), and Stephan Kemp, Ph.D.
Definition
Adrenoleukodystrophy (ALD) is a genetic disorder that affects the adrenal glands, spinal cord, and the white matter of the brain. It is severe and progressive. The disorder was first identified in 1923 and was formerly known as Schilder’s disease and sudanophilic leukodystrophy. In the 1970s, the name adrenoleukodystrophy was introduced to better describe its manifestations. The term ‘adreno’ refers to the adrenal glands, ‘leuko’ refers to the white matter of the brain, and ‘dystrophy’ means abnormal growth or development. It is important to note that this condition is distinct from ‘neonatal adrenoleukodystrophy,’ which is a disorder in the Zellweger spectrum of peroxisomal biogenesis disorders.
Biochemistry
ALD is a genetic metabolic storage disorder caused by a deficiency in a specific enzyme. This deficiency leads to the accumulation of harmful very long-chain fatty acids (VLCFA) in various tissues. The brain, spinal cord, testes, and adrenal glands are primarily affected. The accumulation of VLCFA damages the myelin sheath around nerves in the central nervous system, causing neurological problems. High levels of VLCFA negatively affect adrenal cells, contributing to Addison’s disease (adrenal insufficiency).

Figure 1: The excess VLCFA that accumulates in ALD is primarily due to the elongation of long-chain fatty acids. To maintain a healthy balance, cells must break down these excess VLCFA. This breakdown process takes place in peroxisomes. ALD is caused by gene mutations in ABCD1, which produces the ALD protein (ALDP). ALDP is responsible for transporting VLCFA into the peroxisomes. When ALDP is deficient, this transport is impaired, leading to an accumulation of VLCFA in cells, tissues and organs. The enzymes needed to break down VLCFA are present in peroxisomes. However, because of the transport blockage, the VLCFA cannot reach them.
Epidemiology
ALD is a global disease that affects people of all ethnicities and regions. It is estimated to affect approximately 1 in 15,000 newborns worldwide..
Genetics
ALD is a disorder caused by a defective gene called ABCD1 located on the X chromosome.
In males (XY), symptoms of ALD occur when the X chromosome carries the faulty gene. If a father carries the faulty gene, there isn’t an extra X chromosome for protection, resulting in symptoms.
Females (XX), who have two X chromosomes, have traditionally been called “carriers,” because it was assumed that only a small percentage would develop symptoms. However, this belief has changed. Although symptoms in females are typically less severe than in males, 80% of females with adrenoleukodystrophy will eventually develop symptoms. Therefore, the term “adrenoleukodystrophy carriers” is now considered misleading and it is recommended that it not be used. It is thought that the presence of a normal ABCD1 gene on an X chromosome explains the milder symptoms in females and protects them from the cerebral variant, which affects the brain. 
Figure 2: Possible outcomes for each newborn: For affected females (left side of Figure 2): If an affected woman has a daughter, there is a 50% chance she will inherit the defective gene and a 50% chance she won’t be affected. If a son is born, there’s a 50% chance that he will have ALD, and a 50% chance that he will be unaffected.
For affected males (right side of Figure 2): Affected men pass their Y chromosome to all their sons, making them free of the disease. However, all daughters will inherit the defective X chromosome.
Clinical course
Individuals with ALD have no symptoms at birth. The clinical course is different in males and females.
In males
In males, the first noticeable symptom of ALD is often adrenal insufficiency, which can occur in infancy. Cerebral ALD, which is progressive cerebral demyelination, can develop in childhood or adulthood.
It may occur as the first sign of ALD or in association with adrenal insufficiency and/or myelopathy (see Figure 3). As men age, myelopathy, a spinal cord disease, becomes apparent.
 Figure 3: This illustration shows the progression of ALD in males, highlighting ages at which different symptoms may begin. The blue bar represents adrenal insufficiency, which can begin as early as 5 months of age. The mauve bar represents myelopathy, a chronic spinal cord disease that develops in adulthood. The green bar represents cerebral ALD, which can occur at any age, with the youngest reported case being 3 years old. The ALD gene defect and storage of VLCFAs cause adrenal insufficiency and myelopathy, collectively known as adrenomyeloneuropathy. The onset of cerebral ALD is likely influenced by a combination of the primary gene defect, unknown environmental triggers, and/or genetic factors. Patients with adrenal insufficiency and/or myelopathy are at risk for developing cerebral ALD.
Figure 3: This illustration shows the progression of ALD in males, highlighting ages at which different symptoms may begin. The blue bar represents adrenal insufficiency, which can begin as early as 5 months of age. The mauve bar represents myelopathy, a chronic spinal cord disease that develops in adulthood. The green bar represents cerebral ALD, which can occur at any age, with the youngest reported case being 3 years old. The ALD gene defect and storage of VLCFAs cause adrenal insufficiency and myelopathy, collectively known as adrenomyeloneuropathy. The onset of cerebral ALD is likely influenced by a combination of the primary gene defect, unknown environmental triggers, and/or genetic factors. Patients with adrenal insufficiency and/or myelopathy are at risk for developing cerebral ALD.
Adrenal insufficiency
Boys and men with ALD may experience adrenal insufficiency and potentially life-threatening Addisonian crises as their first symptoms, which can occur years or even decades before neurological symptoms develop. A study of boys who were pre-symptomatic for neurological symptoms found that 80% had adrenal insufficiency when they were diagnosed with ALD. Common signs of adrenal insufficiency include chronic fatigue, muscle weakness, loss of appetite, weight loss, abdominal pain, unexplained vomiting, nausea, diarrhea, low blood pressure (especially when standing, which can lead to dizziness or fainting), irritability, depression, cravings for salty foods, low blood sugar, headaches, and sweating.
Individuals may or may not have increased skin pigmentation due to excessive secretion of adrenocorticotropin hormone (ACTH).
Myelopathy
Most male patients with ALD who reach adulthood will eventually develop myelopathy. This typically occurs between the ages of 20 and 40. Symptoms primarily affect the spinal cord and peripheral nerves.
Characteristics:
Neurological disability is initially slowly progressive.
Diagnosis is rarely made during the first 3 to 5 years of symptoms unless other familial cases are identified.
Symptoms include slowly progressive gait disturbances due to leg stiffness and weakness.
Bladder dysfunction with urgency may develop, which may progress to complete incontinence.
Symptoms typically worsen over several years or decades, and most patients lose the ability to walk unassisted by their fifth or sixth decade.
Adrenomyeloneuropathy (AMN):
The term adrenomyeloneuropathy refers to male patients with both adrenal insufficiency and myelopathy.
Cerebral ALD
Boys and men with ALD may develop demyelinating lesions in the white matter of the brain, known as cerebral ALD. Recent observations suggest that the prevalence rates in adolescence and adulthood may be higher than previously thought, although onset has not been reported before the age of 3 years.
Historical perspective:
Traditionally, cerebral ALD was thought to be rare in adolescence (4-7%) and adulthood (2-5%). However, systematic MRI studies of a large group of males with ALD now suggest a potentially higher prevalence.
Risk factors and triggers:
Cerebral ALD is unpredictable in timing and onset. While head trauma has been reported as one of the possible triggers, other as yet unknown genetic and environmental factors are likely to play an important role.
Symptoms and progression:
Symptoms of cerebral ALD typically progress rapidly.
Males born with the disease have a 35 to 40% chance of developing cerebral ALD between the ages of 3 and 18.
Early symptoms of ALD in elementary school-aged boys include behavioral problems and learning disabilities. These symptoms are often misattributed to other disorders such as ADHD, which may delay the diagnosis of ALD.
In adults, initial symptoms may mimic depression or psychosis, which can also delay diagnosis, especially if in the absence of a family history or symptoms of adrenal insufficiency.
The progression of the disease is rapid at this stage, with patients losing their ability to understand speech and walk within months.
Eventually, patients become bedridden, blind, unable to speak or respond, and require full-time nursing care and tube feeding.
Patients with this disease usually die within 2 to 4 years of the onset of symptoms. However, with proper care, some patients can remain in a vegetative state for longer periods of time.
Women with ALD
It is now well established that more than 80% of women who carry the defective ALD gene will develop symptoms by the age of 60, contrary to the initial assumption that they will remain asymptomatic. Key points to consider.
Onset and progression:
Neurological symptoms in women typically begin at a later age (between 40 and 50 years) than in men with myelopathy.
Disease progression is generally slower than in males.
Distinctive feature:
Unlike males, fecal incontinence is a common complaint in females with ALD.
Risk of misdiagnosis:
Women with ALD often have their myelopathy misdiagnosed as multiple sclerosis
Adrenal insufficiency and cerebral ALD are rare:
Adrenal insufficiency and cerebral ALD are both very rare in women, occurring in less than 1% of cases each.
Research papers:
Two research papers describing the signs and symptoms in women with ALD are available for free download in (PDF) format.
Please see (Females with ALD) for more information.
Testing
Diagnosis of ALD involves a simple blood test that measures very long-chain fatty acid (VLCFA) levels, which is highly accurate in men of all ages. However, approximately 15% of women with ALD may have normal VLCFA levels, resulting in a “false negative” result.
VLCFA test accuracy:
The blood test for VLCFA is widely accepted and accurate in diagnosing males with ALD.
False negatives in women:
In approximately 15% of women with ALD, the VLCFA test may show normal levels, resulting in a “false negative” result.
DNA testing for women:
DNA testing is recommended for accurate identification of females with ALD, especially those with normal VLCFA levels.
Normal DNA test results reassure females that they are not carriers of the defective adrenoleukodystrophy gene.
C26:0-lysoPC as a diagnostic biomarker:
In 2020, C26:0-lysoPC levels were found to be elevated in all men and more than 99% of women with ALD [Jaspers et al 2020].
Even in women with ALD with normal VLCFA levels, C26:0-lysoPC levels were elevated in dried blood spots and plasma.
C26:0-lysoPC is considered a superior diagnostic biomarker for ALD compared to VLCFA analysis.
Newborn screening
Early diagnosis is critical to saving lives in ALD, and newborn screening plays a pivotal role in enabling prospective monitoring of adrenal function and the onset of cerebral ALD.
Key points include:
Newborn screening test:
A newborn screening test has been developed to detect elevated levels of VLCFA, specifically C26:0-lysoPC, in blood spots.
Introduction in New York:
On December 30, 2013, the State of New York began screening newborns for ALD.
United States Recommended Uniform Screening Panel (RUSP):
In February 2016, ALD was added to the United States Recommended Uniform Screening Panel (RUSP).
Global adoption:
Following the inclusion in the RUSP, other states and countries have initiated newborn screening programs or processes to add ALD to existing programs.
Taiwan initiated ALD newborn screening in January 2016.
The Netherlands began ALD newborn screening on October 1, 2023.
Regional pilots are underway in Italy, Israel, Japan and Spain.
Information resource:
Detailed and up-to-date information on ALD newborn screening can be found on the dedicated page titled Newborn screening”.
Treatment
There is currently no curative treatment for ALD. Various approaches have been explored to treat specific aspects of the disease:
Adrenal steroid replacement therapy
Adrenal insufficiency is common in ALD and is often the first manifestation.
Adrenal steroid replacement therapy is mandatory and can be life-saving, but it doesn’t affect neurological symptoms.
Treatment of Myelopathy
There is no curative therapy for myelopathy, which affects 85% of ALD patients (men and women combined).
Dietary restriction
Because very long-chain fatty acids (VLCFA) can be harmful to myelin, adrenal glands, and testes, various attempts have been made to reduce plasma concentrations of VLCFA. However, limiting VLCFA intake by dietary restriction alone does not affect plasma VLCFA levels.
Lorenzo’s oil
Lorenzo’s oil, a combination of oleic acid and erucic acid triglycerides, was thought to be promising.
However, clinical trials showed that it did not improve neurological or endocrine function or halt disease progression.
For more details, please visit the (Lorenzo’s oil) page.
Lovastatin
Lovastatin was initially considered as a VLCFA-lowering therapy, but conflicting results emerged.
Clinical trials showed a small reduction in plasma VLCFA but no effect at the cellular level.
For more information, please visit the (Lovastatin) page.
Bezafibrate
In the search for compounds that could potentially reduce VLCFA levels, bezafibrate, a drug commonly used to treat hyperlipidemia, was identified as a VLCFA-lowering agent. Experiments in fibroblasts showed that bezafibrate reduced VLCFA levels by directly inhibiting the activity of the VLCFA-specific elongase ELOVL1. To evaluate the effect of bezafibrate on VLCFA accumulation in blood cells from ALD patients, an open-label pilot study was conducted. Unfortunately, the study showed that bezafibrate did not effectively reduce VLCFA levels in the blood cells of ALD patients. This lack of efficacy is likely due to the inability to achieve sufficient drug levels in patients.
Bone marrow transplant (HSCT)
In boys and adolescents with the early stages of cerebral ALD, allogeneic hematopoietic stem cell transplantation (HSCT) has the potential to halt the progression of cerebral demyelination. This positive outcome depends on the procedure being performed at a very early stage of the disease. The efficacy of HSCT is based on the replacement of ALDP-deficient brain microglial cells with normal microglial cells derived from donor bone marrow stem cells.
For more details, please visit the (Hematopoietic stem cell transplantation) page.
Gene therapy
Transplantation of autologous hematopoietic stem cells (the patient’s own bone marrow cells) genetically corrected ex vivo (outside the patient’s body) with a lentiviral vector prior to reinfusion is emerging as an additional therapeutic option. This optimism stems is based on the encouraging results reported in 2009 from the treatment of the first two ALD patients, as well as the more recent data from the Starbeam Study published in October 2017.
For more details, please visit the (Gene Therapy for ALD) page.
A 10 minute overview of adrenoleukodystrophy
Produced by Youreka Science in collaboration with ALD Connect, Inc.
Please see the ALD Connect Educational Videos & Webinars page for more videos.
Newborn screening
Introduction
Babies born with adrenoleukodystrophy (ALD) are neurologically normal at birth. However, early diagnosis of boys with ALD can lead to life-saving interventions. These include the timely initiation of adrenal steroid replacement therapy after identification of adrenal insufficiency and the provision of allogeneic hematopoietic stem cell transplantation (HSCT) as a means of treating cerebral ALD. HSCT can halt the often fatal progression of cerebral demyelination, provided the procedure is performed at a very early stage of the disease. Unfortunately, this can only be effective during a narrow therapeutic window that is often missed. Newborn screening provides access to this “window of opportunity” and allows timely initiation of these established therapies.
In February 2016, ALD was added to the US Recommended Uniform Screening Panel (RUSP). This is the federal list of all genetic conditions recommended for state newborn screening programs. New York State initiated newborn screening for ALD on December 30, 2013. Since then, the majority of U.S. states have initiated newborn screening for ALD (Fig 1). As of 2024, ALD screening is available in most states, with additional states continuing to implement programs.
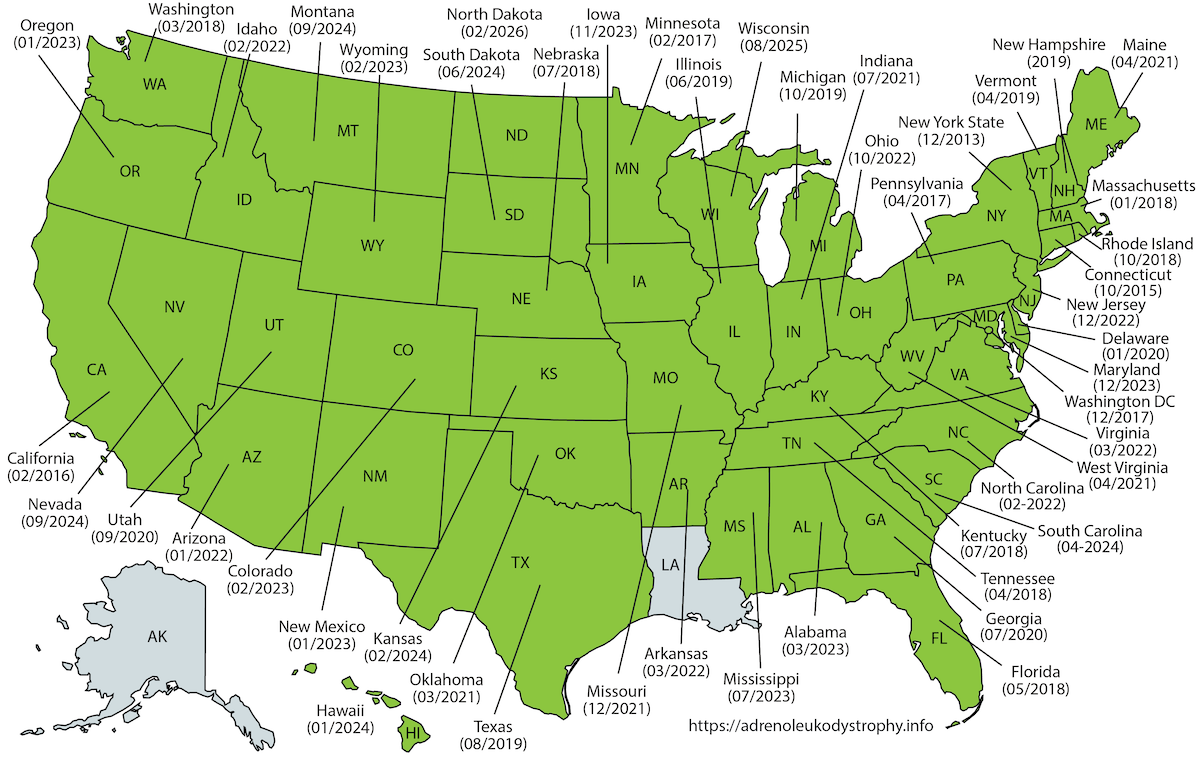
Figure 1: Map showing the states in the US that have initiated ALD newborn screening.
Outside the United States, Taiwan initiated newborn screening for ALD in November 2016 (Chen et al. 2022). Pilot programs and implementation efforts are underway in Japan (Shimozawa et al. 2021), Israel, Italy (Bonaventura et al. 2023) and Spain. Following a successful pilot study in 2021 (Albersen et al. 2022), the Netherlands initiated nationwide newborn screening for ALD on October 1, 2023. The Netherlands is the first country to implement sex-specific screening, screening only males who are at high risk for adrenal insufficiency and cerebral ALD during childhood. Boys with ALD are identified through a combination of elevated C26:0-LPC levels, sex determination (the X-counter), and confirmation of a pathogenic ABCD1 variant (Barendsen et al. 2020). Several other countries are evaluating ALD for inclusion in their screening programs.
Criteria for inclusion in the screening program
There is broad international consensus on the criteria for inclusion of a condition in a newborn screening program.
- Early diagnosis must be of direct benefit to the newborn. There must be a substantial health benefit to be gained from early intervention in serious diseases with a known natural history.
- The screening test must be of good quality. The test must have high specificity and sensitivity, i.e. a very low rate of false positive and false negative results.
 History
History
In 2004, at a meeting of the National Advisory Committee for Newborn Screening, Dr. Hugo Moser suggested that ALD be added to the U.S. RUSP. The only problem was that there was no valid test for newborn screening. To overcome this, he raised funds and recruited a team of researchers at the Kennedy Krieger Institute (Baltimore, MD) to identify a suitable biomarker and develop a test using tandem mass spectrometry (MS/MS). In 2006, the team reported the identification of C26:0-lysophosphatidylcholine (C26:0-LPC) in postnatal venous dried blood spots (DBS) from ALD males (Hubbard et al. 2006). In the following years, scientists continued to improve the analysis (Hubbard et al. 2009; Theda et al. 2014). Then, in collaboration with researchers at the Mayo Clinic (Rochester, Minnesota), a high-throughput method for the analysis of C26:0-LPC was developed (Haynes and De Jesús 2012; Turgeon et al. 2015). In 2013, this method was validated using 100,000 anonymous dried blood spots.
 Aidan’s Law
Aidan’s Law
In April 2012, after the death of their son, Aidan, who had cerebral ALD but was diagnosed too late, the Seeger family drafted and supported the passage of Aidan’s Law in New York State. The bill passed in February 2013 and became law in March 2013. On December 30, 2013, the New York State Newborn Screening Laboratory began testing babies for ALD.
New York State
In the first three years, New York State screened over 700,000 newborns and identified 45 babies with ALD: 22 boys and 23 girls (Moser et al. 2016). Based on these numbers, the birth incidence of ALD is 1 in 15,000. When a newborn with ALD is identified, the family is referred to a clinical geneticist for confirmation of the diagnosis, along with genetic counseling for support services and screening of other family members at risk for ALD (extended family screening).
In males, it is imperative to initiate serial monitoring with brain MRI to detect the earliest signs of onset of cerebral ALD and to initiate adrenal function testing to detect adrenal insufficiency. A comprehensive evaluation of neurological, neuropsychological, neuroradiological, and adrenal function is necessary because there is no test to predict the clinical outcome of an individual child born with an ALD pathogenic variant.
The newborn screening test
The details of the C26:0-LPC test may vary slightly from state/country to state/country. In general, the diagnosis of ALD is made using a three-step (tier) algorithm (Fig 2). The first step is a high-throughput standard MS/MS analysis of C26:0-LPC. Samples with elevated levels of C26:0-LPC are then screened in the second step using HPLC-MS/MS. This test is more sensitive, but also a little more time consuming. For those samples that still show elevated C26:0-LPC, sequencing of the ABCD1 gene is performed in the third step.
 Figure 2: The principles of ALD 3-tier screening.
Figure 2: The principles of ALD 3-tier screening.
Challenges: Screening boys and girls, or boys only?
There are significant challenges and ongoing ethical discussions in different countries regarding the implementation of newborn screening for ALD.
- A fundamental question in ALD newborn screening is whether to screen all newborns or only males. This debate centers on the first criterion for inclusion: early diagnosis must be of direct benefit to the newborn.
- In males, approximately one third will develop cerebral ALD between the ages of 3 and 18 years, and approximately 50% will develop adrenal insufficiency, both of which can be effectively treated when detected early. In adulthood, virtually all males will develop myeloneuropathy in adulthood, which is characterized by limb spasticity, gait dysfunction, and incontinence. Myeloneuropathy is treated symptomatically, and there is currently no disease-modifying therapy.
- Females with ALD have a <1% chance of developing adrenal insufficiency or cerebral ALD during childhood. So, newborn screening offers no direct health benefit for girls in terms of treatable childhood conditions. Approximately 80% of women with ALD will develop myelopathy by the age of 60, but like males with myeloneuropathy, this is not a condition that benefits from early childhood detection.
- Some countries, like the United States, Italy, Israel and Spain screen all newborns and identify both boys and girls. Others, like Japan and the Netherlands, have chosen to screen only males who are at risk for childhood-onset treatable conditions. Both approaches have merit, and the choice reflects differing interpretations of screening criteria and ethical frameworks.
Variant Classification and the Grey Zone
Newborn screening for ALD frequently identifies genetic variants of uncertain significance (VUS) in the ABCD1 gene. These variants are associated with borderline-elevated C26:0-LPC levels, creating diagnostic uncertainty for families. Traditional pathogenicity classifications do not adequately account for age-dependent disease penetrance or the variable clinical spectrum of ALD.
To address this challenge, risk-stratification frameworks have been developed that integrate biochemical data (C26:0-LPC levels) with genetic and clinical information. These frameworks classify variants into risk categories such as “no ALD,” “lower-risk for childhood disease,” and “at-risk for childhood disease.” This approach helps distinguish true positives from false positives, reduces unnecessary medical interventions, and provides families with evidence-based guidance about actual disease risk.
International collaboration through initiatives like the Grey Zone Project and the ABCD1 Variant Registry (adrenoleukodystrophy.info) continues to refine variant interpretation and improve screening accuracy worldwide.
Recommendations for management
International recommendations for the diagnosis and management of patients with adrenoleukodystrophy: A consensus-based approach
In 2022, a consensus-based modified Delphi approach, a structured communication technique used to reach consensus among a panel of experts through multiple rounds of questionnaires and feedback, was conducted among 28 international ALD experts to develop best practice recommendations for the diagnosis, clinical surveillance, and management of patients with ALD.
 Figure 1: The Delphi approach consisted of two rounds of questionnaires and a consensus meeting. In the first round of questionnaire, experts rated their agreement with statements derived from the clinical questions on a 9-point scale (1: “strongly disagree” and 9: “strongly agree”). Consensus was defined as more than 80% of the experts rating their agreement between 7 and 9 or their disagreement between 1 and 3.
Figure 1: The Delphi approach consisted of two rounds of questionnaires and a consensus meeting. In the first round of questionnaire, experts rated their agreement with statements derived from the clinical questions on a 9-point scale (1: “strongly disagree” and 9: “strongly agree”). Consensus was defined as more than 80% of the experts rating their agreement between 7 and 9 or their disagreement between 1 and 3.
The experts identified 39 discrete areas of consensus, including screening, treatment and management of cerebral ALD, myeloneuropathy, adrenal insufficiency, and dietary therapy, addressing a clinical need in the ALD community worldwide as the number of diagnoses and presymptomatic individuals increases due to newborn screening and the greater availability of next-generation sequencing. The poor ability to predict disease course and progression informs current surveillance intervals but is subject to change as more data become available. This knowledge gap should guide future research and once again illustrates that international collaboration among physicians, researchers, and patients is essential to improve care.
The full publication in Neurology is freely available.
 Figure 2: Overview of the Management of Patients With ALD.
Figure 2: Overview of the Management of Patients With ALD.
Variants in ABCD1
The ABCD1 Variant Registry reports all variants in accordance with the nomenclature recommended by the Human Genome Variation Society. All variants, including those that have been published in the past, are annotated using Alamut software. The transcript NM_000033.3 on GRCh37 (hg19) is used as the reference sequence. The ABCD1 Variant Registry is a community-driven project. Its strength lies in its collaborative nature, with diagnostic laboratories, researchers, and physicians able to contribute new variants and updates regarding pathogenicity. If you use the ABCD1 Variant Registry as a reference guide, then please share ABCD1 variants and/or updates regarding pathogenicity with the ABCD1 Variant Registry. This will help us to continuously make improvements (your contribution will be acknowledged).
Because ABCD1 pathogenic variants have no predictive value with respect to the clinical outcome of an individual patient, no phenotypic information is provided. Instead, we report cases. An ALD case is defined as an individual who has been diagnosed with clinical signs and symptoms related to ALD (adrenal disease, myeloneuropathy and/or cerebral ALD), and a biochemical or genetic confirmation. Where available in the scientific literature, experimental data were extracted supporting the pathogenicity of a particular variant. The registry has been expanded to include variants identified through ALD newborn screening programs. For these variants, biochemical data (C26:0-lysoPC levels) are incorporated as part of the pathogenicity assessment, and dedicated classification categories have been developed to reflect the unique context of newborn screening.
Go to The ABCD1 Variant Registry